

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Vasospastic angina (VA), also called Prinzmetal's angina or variant angina, is a specific type of coronary artery disease and is developed due to not atherosclerosis but coronary artery spasm. This disease can cause acute coronary syndrome, including acute myocardial infarction, ventricular tachycardia, fibrillation or electro mechanical dissociation, and even sudden cardiac death.1) The incidence of VA is higher in Asians than Caucasians or Blacks,2)3) which strongly suggests that genetic traits might be involved in the pathogenesis of VA.4-11)
Studies have shown that the loss of Kir6.1 {poreforming inwardly rectifying K+ (Kir) channel subunit} or SUR2 (sulfo-nylurea receptor regulatory subunit of KATP channel) by related gene deletions in mice induced VA.4)5) So far, only these two genes have been shown to be associated with gene copy number alterations. Other investigations showed gene polymorphisms6-8) and functional expression9-11) in VA. Related gene polymorphisms in VA included an endothelial nitric oxide synthase gene,6) adrenergic receptor gene,7) and the paraoxonase 1 gene.8) The Rho-associated kinase gene9) and serotonin receptor (5-HT1Dβ) gene10)11) were also shown to be related to VA by their functional expression. To date, there have been no studies on deoxyribonucleic acid (DNA) copy number alterations in the whole human genome among patients with VA. The recent development of array-comparative genomic hybridization (CGH) provides a means to quantitatively measure DNA copy number aberrations and to map them directly to genomic sequences.12-14) Also, tiling resolution DNA microarrays comprised of large-insert genomic clones such as bacterial artificial chromosomes (BACs) are a very useful and accurate method for detecting genomic aberrations in many genetic diseases.15)16) Furthermore, array-CGH with BAC clones, has been proven to be a very useful and accurate tool for detecting genomic aberrations in whole blood samples for schizophrenia, epilepsy and mental retardation,17-19) as well as in tissue from some cancers.20-24) Both fluorescence in situ hybridization (FISH) and real-time polymerase chain reaction (RT-PCR) have also been applied empirically to confirm array-CGH results.17)18)21-24)
In the study presented here, we analyzed genomic DNA copy number aberrations in whole blood from 28 patients with VA using array CGH. Genetic aberrations, for a subset of gained or lost gene, were confirmed by RT-PCR.
Subjects and Methods
Clinical description
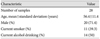
Peripheral blood samples were collected from 28 patients with VA at the Department of Cardiology at Seoul St. Mary's Hospital, Seoul, Korea. In this study, the diagnosis of VA was made when patients met all of the following criteria: 1) repetitive burning or squeezing retrosternal chest pain, 2) definite positive response by coronary angiography with intra coronary acetylcholine provocation test, 3) no significant atherosclerotic stenosis in coronary artery (<50% narrowing of the coronary luminal diameter) according to quantitative coronary angiography. Blood samples were collected from 8 women and 20 men, aged 34-71 years (mean age±standard deviation, 56.44±11.4 in years). The clinical characteristics of the patients are shown in Table 1. Genomic DNA was extracted using a Puregene DNA isolation kit (Qiagen, Hilden, Germany). Reference DNA was pooled from 10 gender-matched (male), normal, healthy control subjects. All patients gave written informed consent to participate in this study. The protocol of this study was approved by the institutional review board of Seoul St. Mary's Hospital, The Catholic University of Korea.
Array comparative genomic hybridization
Array CGH analyses were conducted on 28 individual samples using commercial MACArray™ Karyo 4K BAC-chips (Macrogen, Seoul, Korea) with 4,030 BAC clones, in duplicate, on the whole human genome with a resolution of about 1 Mbp. Array CGH was performed as described previously.25) Briefly, 500-700 ng target and reference DNAs were denatured in the presence of random primer and reaction buffer (BioPrime® DNA Labeling System, Invitrogen, Carlsbad, CA, USA) at 98-100℃ for 5 minutes, and then cooled on ice for 5 minutes. The denatured DNA was differentially labeled with 3 µL of 1 mM Cy3- and Cy5-conjugated dCTP by random primed labeling (Perkin Elmer, Waltham, MA, USA). The mixture was incubated with a Klenow fragment at 37℃ overnight. After labeling, unincorporated nucleotides were removed using MicroSpin™ G-50 columns (Amersham Biosciences, Buckinghamshire, UK). Cy3- and Cy5-labeled test DNA and reference DNA were mixed with 50 µL of human Cot-1 DNA for blocking of repeat sequences. After purification, this mixture was resolved in hybridization buffer (Macrogen) containing yeast tRNA for blocking of non-specific nucleotides binding.
After the MACArray™-Karyo 4K BAC-chip was prehybridized in hybridization buffer with salmon sperm DNA for 1 hour, chips were hybridized with the purification mixture. It was then incubated for 72 hours in the 37℃ hybridization chamber (BioMicro systems, Salt Lake City, UT, USA). After hybridization was complete, array chips were washed in 50% formamide-2x SSC at 46℃ for 15 minutes, and 0.1% SDS-2x SSC at 46℃ for 30 minutes. Next, the chips were washed in 50% sodium phosphate-0.1% NP40 for 15 minutes followed by washing with 2x SSC for 15 minutes at room temperature. After spin drying, hybridized arrays were scanned with a Genepix™ (Axon Instruments, Sunnyvale, CA, USA).
Data analysis
Raw signal intensities from the arrays were measured using the MAC Viewer v1.6.3 software (Macrogen). Log2 (Cy3 intensity/Cy5 intensity ratios) were normalized by using the median of fluorescence ratios computed from the housekeeping DNA control fragments linearly distributed across the genome. Measurements flagged as unreliable by MAC Viewer v1.6.3 were removed from subsequent analysis. The threshold corresponds to 2 standard deviation (SD) values from the mean. The information on each individual clone was obtained from the UCSC Genome Bioinformatics database (May 2004 freeze, http://genome.ucsc.edu).
Reverse transcriptase quantitative polymerase chain reaction
To confirm the level of genomic imbalances identified by array CGH in this study, DNA samples with obvious genomic changes were analyzed using RT quantitative PCR.26) The reaction was performed in a total volume of 50 µL, including 25 µL of 2x IQ™ SYBR® green supermix (Bio-Rad, Hercules, CA, USA), 10 ng of DNA, and 10 pmol of each primer.27) Forty cycles of amplification, data acquisition, and data analysis were performed in an iCycler (Bio-Rad). Primers for eight genes (CTDP1, HDAC10, KCNQ1, NINJ2, NOTCH2, PCSK6, SDHA, and MUC17) were selected and the position of each clone was obtained from the UCSC genome database (Table 2). The relative genomic copy number was calculated using the comparative CT Method.28) GAPDH was used as an endogenous reference.28-30)
Results
Genomic deoxyribonucleic acid copy number changes and pattern of aberrations
Chromosomal copy number changes were detected by array-CGH in VA. Copy number changes were presented as individual chromosome plots of log2 ratio of normalized Cy3 : Cy5 intensities versus chromosome regions for each of the individual 28 cases of VA. Table 3 shows the frequency of aberration, ranging from 7 (25% of chromosomal gain and loss) to 15 (54% of chromosomal loss at 7q) in 28 samples. Overall, the individual chromosomal aberration pattern was not random and tended to be consistent, showing DNA gains in 1p, 1q, 2p, 2q, 3p, 3q, 4p, 4q, 5p, 5q, 6p, 6q, 7q, 8p, 8q, 9q, 10p, 10q, 11p, 11q, 12q, 13q, 14q, 15q, 16p, 16q, 17p, 17q, 18q, 20p, 20q, 21q, and 22q; and showing DNA losses in 2p, 4q, 7q, 10q, 15q, 16p, 17p, and 19p. DNA gains, rather than DNA losses, were more frequently observed. The most frequent gain detected was at 5p15.33, and 11q12.2 (50%). Frequent DNA gains also were observed in 12p13.33, 14q32.33, and 15q11.2 in 13 samples (46%), and in 12p13.33 (43%), 1p12-p11.2, 4p15.1, 8q22.2, 11q25, and 15q26.3 in 11 samples (39%). Frequent gains (36%) were located at: 1q44, 3q24, 11q23.3, 15q11.2, 15q25.2, 15q26.3, 16p12.1, 16p13.2, 17p12, 18q12.3, and 21q11.2. Other regions with 25-32% frequency of DNA gains included 1p12-p11.2, 1p12, 1p21.1, 2p11.2, 2q21.2-q21.3, 2q22.1, 2q23.3, 2q31.1, 2q37.3, 3p14.1, 3q12.3, 4p15.1, 4p15.2, 4q24, 4q32.3, 4q34.3, 4q35.2, 5p12, 5p15.2, 5p15.33, 5q15, 6p12.2, 6q24.1, 7q11.23, 7q22.2, 8p23.3, 8q24.3, 9q22.1, 9q22.32, 9q33.3, 10p15.3, 10q11.23, 10q23.1, 10q26.3, 11p15.5, 11p15.5-p15.4, 11q14.1, 11q23.1, 11q23.2, 12p13.33, 12q22, 13q33.3, 14q31.3, 15q26.3, 16p11.2, 16q24.1, 17q24.3, 17q25.3, 18q23, 20p12.2, 20q12, 20q13.33, 21q11.2, 21q22.3, 22q12.2, and 22q13.33. The most common genomic losses were observed in 7q22.1 (54%), 4q35.2 (43%), and 15q11.2 (39%), 10q26.3 (36%). Other regions with 25-32% frequency of DNA losses included 2p24.1, 7q36.3, 10q26.3, 16p13.11, 16p13.3, 17p11.2, and 19p13.3 (Table 3). Fig. 1 shows chromosomal aberration regions. 1p12-p11.2, 5p15.33, 11p15.5-p15.4, 12p13.33, 15q26.3, 18q23, and 22q13.33 contained CTDP1, HDAC10, KCNQ1, NINJ2, NOTCH2, PCSK6, and SDHA genes (gained regions), while loss region 7q22.1 contained MUC17 gene.
Result according to reverse transcriptase quantitative polymerase chain reaction
To confirm the array CGH results, differential fold change was evaluated by RT quantitative PCR in VA samples. The results showed chromosomal fold changes in CTDP1 (18q23), HDAC10 (22q13.33), KCNQ1 (11p15.5-p15.4), NINJ2 (12p 13.33), NOTCH2 (1p12-p11.2), PCSK6 (15q26.3), SDHA (5p15.33), and MUC17 (7q22.1). Fig. 2 shows the RT PCR results. In gained regions of 7 genes, {CTDP1, HDAC10, KCNQ1, NINJ2, NOTCH2, PCSK6, and SDHA (A-G)}, the relative fold increases in RT quantitative PCR corresponded with array-CGH results. In MUC17 (H), the decrease in fold differences noted by RT quantitative PCR was similar to that seen with array-CGH. The N-value was delineated in RT PCR. The results compared linear-ratios in array-CGH, were demonstrated generally larger by RT quantitative PCR analysis than those by array-CGH analysis.
Discussion
Several methods including classical cytogenetics, FISH, Southern blot analysis, metaphase CGH, and array CGH have been used to detect DNA copy number changes.23) Lately, array CGH has been shown to be a powerful tool for detecting genetic aberrations and genetic mapping, and offers high resolution, high-throughput, accuracy, and sensitivity for genetic analysis.12-14) It also allows the simultaneous quantitative analysis of all regions of a large genome,25) and can provide quantitative information at the level of chromosomal gain or loss.21)
Although the confirmation of array-CGH results is usually performed by FISH analysis, this technique has several limitations. For example, evaluation of the full extent of genetic gains or losses in the genome requires obtaining metaphase DNA and interpretion of karyotypes that are often complex and require prior knowledge or markers of sites of interest.2) Many recent studies have shown that RT quantitative PCR instead of FISH is also a very useful and accurate technique for the confirmation of array-CGH results,17)18)21-24) thus making it particularly attractive technique for the identification of acquired genetic aberrations in whole blood from patients with VA. Therefore, we used RT quantitative PCR for the confirmation and quantification of the identified genomic aberrations.
In this study, using array CGH, we have showed than there are genetic aberrations and altered genes related to VA. The DNA aberrations had a tendency to be located at rather specific chromosomal regions. Ulike amplification of oncogenes or deletions of tumor suppressor genes21-24) there were no specific genes amplified or deleted in VA in our study. However, many DNA copy number aberrations (91 DNA gains and 12 DNA losses) were detected. These DNA copy number aberrations were novel and different from those previously detected in the Korean population (DNA gains in 6p21.2 and DNA loses in 1p36.31, 4q13.1-4q22.13, 16p12.1, 21q22.3, and 22q11.22).31) This showed that many genetic alterations might be associated with the pathogenesis of VA.
Because array CGH shows genetic aberrations at the genomic DNA level and not at the messenger RNA or protein levels, many chromosomal regions which might be related to the pathogenesis of VA were found in this study. For example, the KCNQ1 gene, one of the genes analyzed by RT quantitative PCR,27) is translated into the potassium voltage-gated channel subfamily KQT member1, which is probably related to cardiac repolarization (www.ihop-net.org/UniPub/iHOP). Also, the NOTCH2 gene, another one of the genes analyzed by RT quantitative PCR, is translated into neurogenic locus notch homolog protein 2 precursors; notch homolog protein 2 is a receptor for the membrane-bound ligands involved in regulation of cell-fate determination (www.ihop-net.org/UniPub/iHOP). However, the function of the proteins translated by these genes has not been completely clarified, so definite pathogenesis of VA cannot be established based on our results. Nevertheless, our data may become the cornerstone that will establish the function of each genetic aberration involved in the pathogenesis of VA.
Conclusion
In summary, using array CGH, we analyzed genome-wide chromosomal aberrations and screened chromosomal candidate regions related to the pathogenesis of VA. The array-CGH results were confirmed by RT quantitative PCR. This study is the first for searching whole genomic alterations by array CGH in patients with VA. Our study may contribute to further clarification of important chromosomal regions, identification of candidate genes, and understanding of the pathogenesis of VA.




 XML Download
XML Download