

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
A pulmonary arteriovenous malformation (PAVM) is a rare pulmonary vascular anomaly. Greater than 80% of PAVMs are congenital, and approximately 50% of are associated with hereditary hemorrhagic telangiectasia (HHT). In addition to common complaints, such as dyspnea and epistaxis, a PAVM can cause hemoptysis, hemothorax, and serious neurologic complications, such as stroke, seizures, and brain abscesses. Ebstein's anomaly (EA) is a congenital cardiac malformation characterized by downward displacement of the attachment of the septal and posterior leaflets of the tricuspid valve. EA is also a rare disorder, and patients with EA may have various additional cardiovascular anomalies. Although the causes of a PAVM and EA have not been established, embryogenic or genetic factors might give some contributions. However, there are no reports concerning the co-existence of PAVMs or HHT with EA. We present a case of a PAVM with EA that is suspected to have HHT.
Case
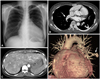
A 40-year-old woman was admitted with a 2-month history of increasing dyspnea. She had experienced spontaneous recurrent epistaxis for the last several years, with no other remarkable medical history. Physical examination of the chest revealed systolic murmurs along the left lower sternal border. A chest radiograph showed cardiomegaly and a well-demarcated, 12-mm nodular opacity in the left lung field (Fig. 1A). The electrocardiogram showed tall and broad P waves, as well as an incomplete right bundle-branch block. On transthoracic echocardiography, the displacement index (distance between the mitral annulus and tricuspid annulus, divided by the body surface area) was 15.6 mm/m2, and it fulfilled the displacement index criteria for the diagnosis of EA (8 mm/m2). Moreover, tethering of the septal and posterior leaflets of the tricuspid valve was observed with central coaptation failure and severe regurgitation. A portion of the right ventricle was atrialized because of apical displacement of the tricuspid valve. The right atrium and ventricle were markedly enlarged (Fig. 2). She was diagnosed with EA, and agreed to undergo surgical treatment. To evaluate the nodule in the left lung, a chest CT angiography was done. A 12-mm serpiginous vascular structure connecting the left interlobar pulmonary artery to the left inferior pulmonary vein was detected in the superior segment of the left lower lobe (Fig. 1B and D). No part of her medical history could explain the causes of secondary PAVM, such as chest trauma, thoracic surgery, long-standing hepatic cirrhosis, metastatic carcinoma, mitral stenosis, or infections. Three-phase contrast CT scans of the liver and MR angiography of the brain were performed to identify other visceral arteriovenous malformations. A liver CT showed diffuse dilatation of the hepatic arteries and veins with multifocal arteriovenous malformations, suggesting HHT (Fig. 1C). The brain MR showed no evidence of vascular malformation. On the 7th day of admission, a tricuspid valve repair was successfully performed without significant complications. After discharge, she has remained stable and comfortable, and will undergo percutaneous embolotherapy of the PAVM in the near future.
Discussion
A PAVM is a rare pulmonary vascular anomaly with an incidence of 2-3 per 100,000 population.1) PAVMs may be single or multiple, and the left lower lobe of the lung is the most common location of solitary PAVM.2) Greater than 80% of PAVMs are congenital, and approximately 50% of these are associated with HHT, also known as Rendu-Osler-Weber disease.2)3) A single PAVM <2 cm in diameter on chest radiography usually does not cause symptoms.4) The most common complaint is epistaxis. A PAVM can cause dyspnea, cyanosis, hemoptysis, hemothorax, and serious neurologic complications, such as strokes, seizures, and brain abscesses.4)5) Patients with co-existing HHT tend to have multiple arteriovenous malformations, rapid disease progression, and a higher complication rate.2) The brain, lungs, and liver are the most frequently involved organs in patients with HHT.6)
EA is a congenital cardiac malformation characterized by downward displacement of the attachment of the septal and posterior leaflets of the tricuspid valve.7) EA is also a rare disorder occurring in 1 per 200,000 live births.7) Most cases are sporadic, and the embryologic and genetic contributors are unclear,8) but the EA gene might be located on chromosome 9.9) The genetic linkages to HHT are located on chromosome 9 or 12.10) Patients with EA may have various additional cardiovascular anomalies.8)11) However, there are no reports concerning the co-existence of a PAVM or HHT with EA. Although it is unclear whether or not a concurrent PAVM and EA has an embryologic or genetic relationship, we report a patient with a PAVM and EA who was suspected to have HHT according to the Curaçao criteria.12) Further genetic and embryonic studies are needed to detect a possible relationship between the two medical conditions.

 XML Download
XML Download