

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
The prevalence of obesity is increasing due to the increasing adoption of westernized life styles in Korea. Obesity is defined by body mass index (BMI), which is classified as underweight (<20 kg/m2, normal (20-24.9 kg/m2, overweight (25-29.9 kg/m2, and obese (≥30 kg/m2. Korean National Health Examination and Nutrition Survey data in 2001 estimated that 3.0% of Korean adults were obese and 29.5% were overweight.1) The prevalence of overweight Korean children and adolescents doubled from 5.4% in 1998 to 11.4% in 2001.2) Increasing degrees of obesity are closely correlated with the increasing rates of cardiovascular disease.3) Furthermore, a large body of evidence strongly supports the view that obesity itself is associated with preclinical structural and functional changes in the heart, which prelude heart failure.4) In this review, we discuss the changes of cardiac structure and function in obesity and focus on the current evidence regarding the causative role of obesity in cardiac changes.
Structural Changes in the Heart in Obesity
Animal studies regarding structural changes of heart in obesity
Two types of animal models are used in obesity studies: genetic mutant models and high fat diet-induced obesity model. Genetic mutant mice models, such as ob/ob and db/db, present severe types of obesity. The ob/ob and db/db mice models become progressively obese, left ventricular (LV) hypertrophy develops, and LV mass increases. Interestingly, leptin infusion to these mice leads to decreased myocardial wall thickness.5) LV hypertrophy is a common cardiac phenotype in the response of genetic mutant mice to obesity. Use of high fat diet mice has demonstrated that increased non-esterified fatty acid (NEFA) contents, which diminish the rate of glucose metabolism and increase oxygen consumption, result in reduced ability to recover from a workload.6) Furthermore, high fat diet mice also display elevated blood pressure and impaired insulin receptor activation.7) All of these factors contribute to the development of cardiac hypertrophy and LV dysfunction in high fat diet mice.7)
Results from the two animal models indicate that obesity may result in cardiac hypertrophy due to insulin resistance (IR), metabolism alteration such as leptin deficiency, or leptin resistance and elevated blood pressure.
Obesity and left ventricular hypertrophy in human
Obesity is an independent factor for LV hypertrophy.8-11) Obesity itself may have hemodynamic effects that produce an increase in the total blood volume and cardiac output due to the high metabolic activity of excessive fat. In moderate to severe obesity, these increases may lead to LV dilation, increased LV wall stress, and compensatory (eccentric) LV hypertrophy.8)9) Despite this logical explanation, recent studies have shown that concentric LV hypertrophy is a predominant form in obese subjects.10)11) Besides hemodynamic factors, hyperinsulinemia due to IR, which stimulates insulin like growth factor-1 (IGF-1) receptors, is also involved in the pathogenesis of LV hypertrophy.12) IGF-1 enhances anabolic effects on the myocardium, and facilitates increased myocardial mass and concentric hypertrophy. Systolic hypertension is traditionally known as a major contributor to LV hypertrophy rather than obesity. However, in the Framingham heart study, the investigators found independent influences of BMI and blood pressure on LV mass index.13) Therefore, the effects of obesity and blood pressure were additive on LV changes. However, opinion is divided concerning which components are the stronger predictor of LV hypertrophy. Obesity was the strongest clinical predictor for LV hypertrophy in several studies,12)14) while another study showed that BMI and hypertension affect LV hypertrophy similarly.15)
In summary, obesity induces LV hypertrophy via hemodynamic changes caused by hyperinsulinemia, increased IGF-1 expression, and volume overload, which are similar to the effect caused by hypertension.
Obesity and cardiac adipocytes in human
Epicardial fat mass, as determined by echocardiographic and magnetic resonance imaging studies, may reflect intra-abdominal visceral fat. Therefore, epicardial fat mass could serve as a marker of visceral adiposity.16) The relationship between local adipose tissue in heart and cardiac geometry has recently been studied. Increased epicardial fat mass and fatty infiltration of myocardium contributed to the increased cardiac mass.17) Although epicardial fat is normally accounted for about 20% of the total ventricular mass, total epicardial fat weights are significantly greater in hypertrophic heart.18)
The causal effect of epicardial adipose tissue on cardiac remodeling remains unsolved. However, epicardial adipose tissue serves as an endocrine organ, which releases monocyte chemotactic protein-1, interleukin-1β, interleukin-6 and tumor necrosis factor-alpha. These adipokines may be important factors in cardiac remodeling or hypertrophy in obese subjects.19)
Obesity and left atrial enlargement in human
Left atrial (LA) size and LA volume is increased in the setting of obesity compared with non-obese individuals.20) The possible mechanisms of increased LA size and volume in obesity are similar to those of LV hypertrophy in obesity; increased BMI leads to volume overload,21) which causes diastolic abnormality of LV filling defect. Diastolic dysfunction can contribute to LA enlargement or remodeling.22) In the Framingham heart study, obesity was found to be a strong risk factor for development of atrial fibrillation even after accounting for concomitant conditions such as hypertension, diabetes mellitus, and myocardial infaction.23) In the clinical setting, dilated LA size in the absence of organic heart disease or atrial fibrillation is considered to be a risk factor for developing atrial fibrillation and long-term cardiovascular events.24)
Functional Changes of the Heart in Obesity
Changes of left ventricular systolic function
The effects of obesity on LV systolic function are controversial. Several studies reported that LV systolic function is normal or increased in obesity.8)9)25) However, some evidence from non-invasive or invasive techniques suggests that obesity causes a subclinical contractility abnormality.11)26)27) Obesity-induced myocardial dysfunction can be explained by the derangement of myocardial metabolism. In animal models, IR in obesity cause alterations in myocardial fatty acid metabolism and efficiency (cardiac work/myocardial oxygen consumption) that occur early in the cascade of events, leading to impaired LV contractility.28) In human studies, obesity is a significant predictor of increased myocardial oxygen consumption and decreased efficiency, and IR is a robust predictor of fatty acid uptake, utilization, and oxidation. These metabolic changes may play a role in the pathogenesis of decreased cardiac performance in obesity.29) Collectively, obesity might be a strong factor that can induce LV systolic dysfunction and eventually cause heart failure independent to coronary artery disease or other morbidities.
Changes of left ventricular diastolic function
Echocardiography studies conducted with obese animals and human have provided inconsistent results about E-wave velocity, deceleration time, A-wave velocity.11)30) Prolongation of the isovolumic relaxation time may be the most consistent diastolic abnormality in obesity.31)32) In uncomplicated obese subjects, diastolic dysfunction is caused by hemodynamic and metabolic factors. Hemodynamic changes cause diastolic dysfunction in obese subjects through LV hypertrophy.33) Metabolic factors will be discussed later chapter. Other potential mediators include hormones and cytokines released in association with obesity. Changes of adipokine concentration in serum, such as leptin and adiponectin, are observed in obese subjects, which may also be partly responsible for the LV diastolic dysfunction. Studies with ob/ob mice (which are deficient in leptin) have demonstrated diastolic dysfunction by the changes of myocardial fatty acid and glucose metabolsim.28)34) Similarly, leptin resistance and hyperleptinemia are observed in obese subjects, which may lead to diastolic dysfunction. Circulating total and high-molecular-weight adiponectin are negatively correlated with LV wall thickness and diastolic dysfunction independent of age and metabolic factors.35) In the Otsuka Long-Evans Tokushima Fatty rat model of pre-diabetes, pre-diabetic conditions cause an accumulation of myocardial collagen, leading to interstitial and perivascular fibrosis, which correlates with LV early diastolic dysfunction.36)
Collectively, the evidence indicates that the preclinical diastolic dysfunction of obesity is related to hemodynamic alteration, various adipokines, and myocardial collagen accumulation.
Several Mechanisms that Influence the Structure and Function of the Obese Heart
Sleep apnea in obesity
Obstructive sleep apnea (OSA) is a very common abnormality in obese subjects. The Wisconsin Sleep Cohort Study demonstrated that the risk of development of hypertension in OSA patients is three times higher than non-OSA subjects.39) Likewise, OSA patients demonstrated not only day-time hypertension but night-time hypertension or non-dipper pattern.39) Repetitive episodes of airway obstruction cause hypoxemia and changes of intra-thoracic pressure, which in turn causes sympathetic overactivity. Furthermore, chronic hypoxemia causes injury to myocytes and cardiac extracellular matrix. All of these factors-hypertension, sympathetic overactivity, hypoxemia-lead to ventricular remodeling.40) One study reported that 88% of OSA patients have LV hypertrophy and 64% of those have LA enlargement.41) The authors also demonstrated that the use of continuous positive airway pressure for 6 months could reduce LV hypertrophy.41) All these findings suggest that the severity of nocturnal hypoxemia could be important in the development of LV hypertrophy in obese subjects with OSA.
Changes of cardiac metabolisms, mitochondrial dysfunction and oxidative stress
Energy sources of myocardial metabolism are different from diabetes compared to normal glucose tolerance subjects. In the setting of diabetes or IR, myocardium utilizes much more free-fatty acid and less glucose, which occurs even in the early course of obesity.42) Long-standing caloric excess or obesity activates peroxisome proliferator-activated receptor-α/peroxisome proliferator-activated receptor-γ coactivator signaling, which increases the expression of genes involved in fatty acid oxidation and fatty acid transporters such as FATP1 and CD36.43)44) In human, obesity is also associated with increased rates of fatty acid oxidation, increased myocardial oxygen consumption, and reduced cardiac performance proportionate to the degree of IR and obesity.29) Indeed, normalization of cardiac metabolism by overexpression of a human GLUT4 transgene in db/db mice reverses cardiac dysfunction, which suggests that altered myocardial metabolism contributes to contractile dysfunction in this model.45) It has been demonstrated that mitochondrial oxygen consumption rate and adenosine triphosphate (ATP) generation capacity are reduced in ob/ob mice, and that the protein levels of mitochondrial complexes I, III, V for the oxidative phosphorylation are significantly reduced in db/db mice.46) Because of mitochondrial response in obesity or diabetes, the ratio of ATP generation and oxygen consumption is reduced (mitochondrial uncoupling), which is believed to be a significant factor for declination of cardiac function.46) Mitochondria also represent a major source of superoxide production. In db/db mice, mitochondrial reactive oxygen species generation increases, which can damage myocardial cells.46)
In summary, cardiac metabolic changes, mitochondrial dysfunction, and oxidative stress can cause declination of cardiac function in obese subjects.
Insulin resistance
IR represents problems of insulin receptor, insulin signaling, and genetics. Among these problems, insulin signaling impairment could be the key factor for IR. Impaired insulin-mediated activation of intracellular signaling has been described in ob/ob mice.42) In the animal models, obesity and IR can increase myocardial fatty acid uptake, which causes myocardial fatty acid oxidation and myocardial oxygen consumption. Persistence of this metabolic change causes an imbalance of fatty acid uptake and fatty acid oxidation, leading to accumulation of fatty acid intermediates and ceramide production, which impairs myocardial function and increases apoptosis of myocardocytes.47)48) This phenomenon has also been demonstrated in human.29) However, in Zucker fatty rats treated with the insulin sensitizer thiazolinedione (TZD), myocardial glucose consumption was increased, fatty acid oxidation was diminished, and myocardial injury was reduced.49) In addition, hyperinsulinemia stimulates IGF-1 receptors, which is likely responsible in the pathogenesis of myocardial hypertrophy.12)
Neurohormonal activations
In general, obese subjects have activated sympathetic tone. This leads to concentric LV hypertrophy due to elevated afterload and increased cardiac contractility. Additionally, catecholamine directly affects the myocardium without hemodynamic influence. The renin-angiotensin system also affects the heart via hemodynamic effects or direct signaling in obesity.50) Angiotensinogen secretion and its messenger ribonucleic acid (mRNA) have been detected in fa/fa rat adipocytes and in human.51) Angiotensin is thought to be a contributor to the sympathoexcitation, therefore the rennin-angiotensin system could increase central sympathetic activation and have a role as an additive effect to blood pressure elevation and cardiac remodeling in obese subjects.52)
Changes of extracellular matrix and fibrosis
The compositional changes of the extracellular matrix is an important contributor in cardiac remodeling.53) Serum levels of cardiac collagen synthesis have been significantly associated with IR in normotensive and non-diabetic obese subjects.54) In another study using a rabbit model of obesity, a high-fat diet caused fibrosis in coronary vessels as well as the accumulation of collagen in the cardiac interstitium.55)
Adipokines such as adiponectin or leptin play an important role in the extracellular matrix changes of the heart. Leptin is an adipokine that is produced in the obese gene (ob) located in adipocytes.56) In one study, leptin increased matrix metalloproteinase-2 (MMP-2) secretion and its mRNA expression, and attenuated collagen I mRNA synthesis and increased collagen III and IV, but did not change total collagen synthesis in human pediatric ventricular myocytes.57) In a diet-induced obesity murine model, elevated leptin level was detected after 20 weeks of a high fat diet, and this leptin level was correlated with reduced ventricular shortening and increased LV posterior wall thickness.58) In the coronary ligation model, procollagen I and III levels were elevated after 7 days postinfarction. Furthermore when the neutralizing leptin antibody was injected, enhanced collagen was attenuated.59)
Adiponectin is considered a cardioprotective adipokine. A recent study suggested that adiponectin level showed positive correlation with tissue inhibitor of metalloproteinase (TIMP), which is considered to exert an antifibrotic effect.60) The adiponetin levels are decreased in the obese or IR subjects; the cardioprotective effect of adiponectin, especially its antifibrotic effect, is diminished in obese subjects.
In obese subjects, adipocytokines such as catecholamine, rennin-angiotensin-aldosterone system, tumor necrosis factor-alpha, C-reactive protein, leptin, and adiponectin influence to MMP activity, TIMP expression, and collagen synthesis, which ultimately leads to cardiac remodeling.53)
Apoptosis of cardiomyocytes
The evidence of cardiac apoptosis in the genesis of heart failure has surged over the last decade. Apoptotic cardiomyocyte death has been proven from biopsies of dilated cardiomyopathy and end stage heart failure.61) Besides apoptotic cell death, activation of several proteases in the apoptotic cascade can cleave contractile proteins including actin, myosin, and troponins.62) There are numerous causes of apoptosis. Apoptosis appears to be ischemia or ischemia-reperfusion driven at the site of infarct and at sites remote from the ischemic area through neurohormonal effects. Myocardial stretch, wall stress, cytokines, and neurohormones such as norepinephrine and angiotensin II, which are commonly produced as a part of peripheral neurohormonal rearrangements after an acute myocardial infarction, have been demonstrated to enhance apoptosis.63)64)
Studies for obesity associated cardiac apoptosis are lacking. Use of the Zucker fatty rat model has shown that cardiac contractility is reduced with increased cardiac cell apoptosis; however, when the rats were treated with TZD, cardiac cell apoptosis was decreased.48) Another study involving a high fat diet induced IR rat model did not reveal a significant difference in apoptosis compared with control group, despite impaired cardiac function.65) Therefore, it is necessary to study the relationship between obesity, cardiac cell apoptosis, and impairment of cardiac function.
Therapeutic Implications
Weight reduction improves systolic blood pressure, IR, sleep apnea, and hyperlipidemia. Weight reduction also has beneficial effects on cardiac structure and function such as reduction of LV diameter, LV wall thickness, LV mass, and LA dimension.66) However, removal of subcutaneous fat by liposuction does not produce a beneficial effect on metabolic changes, so it has little effect on the cardiovascular system.67) Pharmacological-mediated weight reduction is recommended for patients in whom lifestyle modification has failed. Oristat, which is a gastrointestinal lipase inhibitor, can reduce weight by about 3 kg and decreases progression to diabetes in high risk patients.68) Sibutramine, a monoamine reuptake inhibitor, can reduce weight by about 4-5 kg and improves serum lipid levels and insulin sensitivity,69) but it increases heart rate and blood pressure. Rimonabant, an endocannabinoid receptor blocker, reduces weight by 4-5 kg, decreases waist circumference, and improves serum high density lipoprotein cholesterol and triglyceride levels.70) However, there are no definitive data for the effect on cardiac structure and function in use of these medications.71) Therefore, weight reduction by diet and exercise is still the most important means to correct the functional and structural deteriorations that occur in the heart in obese individuals.
Conclusions
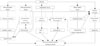
Obesity is a causative factor for development of preclinical changes of the heart. In obese subjects, initially volume overload develops, which leads to increased cardiac output and hypertension. Second, sympathetic and rennin-angiotensin-aldosterone system activity is increased, which causes hypertension. Third, hyperinsulinemia (IR) increases advanced glycation end-products and IGF-1 production and facilitates fatty acid uptake, which leads to lipotoxic cell damage. Fourth, visceral adipose tissue affects oxidative stress and is related with decreased leptin level or leptin resistance and decreased adiponectin level; these changes affect cellular changes and metabolic alteration. Fifth, cellular changes are produced by neurohormonal and metabolic factors, oxidative stress, and adipokines, which leads to extracellular fibrosis and apoptosis. All these descriptive factors eventually lead to structural and functional changes of the heart in obesity (Fig. 1). Therapeutically, life-style associated weight reduction may be more important than any other molecular- or pathway-targeted therapy.
 XML Download
XML Download