

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
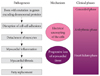
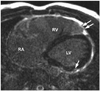
Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) has increasingly been recognized as a common cause of sudden death in young individuals, especially in competitive athletes.1-4) ARVC/D is an inherited myocardial disease characterized histopathologically by progressive loss and fibrofatty replacement of the myocardium. It is typically inherited in an autosomal dominant pattern with variable penetrance. Increasing evidence suggests that genetically determined disruption of desmosomal integrity is a key factor in the development of ARVC/D (Fig. 1).2)3)5) This view has been supported by the identification of mutations in genes encoding desmosomal proteins (desmoplakin, plakoglobin, plakophilin 2, desmocollin 2, and desmoglein 2) in more than 50% of confirmed patients with ARVC/D.6-9) Desmosomal mutations may affect both right and left ventricles.10) Impaired desmosomal function causes separation of myocytes at their intercalated disks and death of myocytes. Myocardial damage from desmosomal dysfunction may be facilitated by mechanical stress such as in endurance exercise.
ARVC/D usually involves the right ventricle preferentially, possibly because the thinner right ventricle is more vulnerable to mechanical stress than the interventricular septum or the left ventricle. It affects predominantly young individuals, particularly athletes.
The natural history of ARVC/D is determined by the electrical instability of the damaged myocardium and the extent of myocardial tissue loss (Fig. 1).10-12) The electrical instability can precipitate arrhythmia, causing cardiac arrest and sudden death any time during the course of the disease, while the progressive myocardial loss causes gradual deterioration of the ventricular function and results in heart failure in the later phase of the disease. Arrhythmias may occur early in the disease before development of significant ventricular remodeling or dysfunction, characterizing ARVC/D differently from other forms of cardiomyopathies, such as dilated and hypertrophic forms.
ARVC/D is a challenging diagnosis. This is especially true in the pediatric population because ARVC/D is an evolving phenotype and the pathological changes are typically subtle in children. Imaging modalities used for the evaluation of right and left ventricular abnormalities in ARVC/D include conventional angiography, echocardiography, computed tomography, and magnetic resonance imaging (MRI). Owing to its ability to visualize the right ventricle clearly and to provide the most accurate measurements of the ventricular volumes and ejection fractions without the use of ionizing radiation, MRI is routinely requested in patients with a confirmed or suspected diagnosis of ARVC/D and in family members of the patients with ARVC/D in most institutions.
At the Hospital for Sick Children in Toronto, ARVC/D is the second most common indication for cardiovascular MR. The purpose of this paper are to summarize the important features of ARVC/D in childhood and compare them with those seen in later life, and to propose a tailored MRI approach to ARVC/D in children.
Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia in Children
As a genetically determined disease, ARVC/D progresses continuously from conception throughout childhood.2-4) The onset of clinical manifestation is typically during adolescence or young adulthood. In a large autopsy series of unexplained sudden cardiac death cases, 13% of deaths from ARVC/D occurred under the age of 18 years.1) Nonetheless, the clinical diagnosis of ARVC/D in children is exceedingly challenging as pediatric patients infrequently meet the Task Force Criteria for Diagnosis of ARVC/D.4) The so-called 'concealed phase' is the period of ARVC/D when there is an early predisposition to ventricular arrhythmia and sudden cardiac death in the context of well-preserved morphology, histology, and ventricular function.13) Most children with ARVC/D would be categorized as being in this phase.
Early identification of the genetically determined but concealed cases is of utmost importance as the sudden cardiac death of the affected patient may be prevented by implementing appropriate and timely medical attention and management.14-16) Corrado et al.14) showed the dramatic impact of a universal screening cardiovascular test based on 12-lead electrocardiography (ECG), history, and physical examination in young competitive athletes. They showed that the annual incidence of sudden cardiovascular death in athletes decreased approximately by 90%, from 3.6/100,000 person-years in the pre-screening period to 0.4/100,000 person-years in the late screening period, and that most of the reduced death rate was due to fewer cases of sudden death from either hypertrophic cardiomyopathy or ARVC/D.
Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia, a Challanging Diagnosis
The diagnosis of ARVC/D can be difficult because of the non-specific nature of the clinical findings, broad spectrum of phenotypic manifestation, and low accuracy of the individual diagnostic tests.2)3)17) Therefore, the diagnosis of ARVC/D has been based on a combination of so-called major and minor criteria proposed by the International Task Force in 1994 (Table 1).18) The criteria include those for structural, functional, electrocardiographic, arrhythmic, genetic, and histological abnormalities. To fulfill the appropriate criteria for ARVC/D, patients must have two major criteria, one major and two minor criteria, or four minor criteria. However, the diagnosis based on these 1994 Task Force criteria has been shown to lack sensitivity for identification of early/minor phenotypes, particularly in the setting of familial ARVC/D. A further limitation of the 1994 criteria was the lack of quantitative cut-off values for dilatation, reduced ejection fraction, and fibrofatty myocardial replacement of the right ventricular (RV). They also did not adjust for differences in normal values within a pediatric population.
The reliability of diagnostic tests is also strongly questioned. A large prospective multicenter study showed considerable discrepancies between the initial and final diagnosis following evaluation of the tests by the core laboratories.17) This study also showed the favorable diagnostic performance of echocardiogram, right ventricular angiogram, signal-averaged electrocardiography (SAECG) and ECG, and inferior performance of MRI and right ventricular biopsy.
Modifications of the original Task Force criteria have recently been proposed (Table 2).19) The revised criteria provide guidance on the role of emerging diagnostic modalities and advances in the genetics of ARVC/D to improve the diagnostic sensitivity with the diagnostic specificity maintained. The revised criteria include quantitative criteria that define the abnormalities on the basis of comparison with normal data, and some of the imaging criteria now adjust for patient size. The quantitative data for global or regional dysfunction and structural alterations are provided by either echocardiography or MRI. The modified Task Force criteria are expected to improve the diagnosis and management of ARVC/D.
A definitive diagnosis of ARVC/D is still based on histological demonstration of transmural fibrofatty replacement of RV myocardium at biopsy, necropsy, or surgery.19) However, the diagnosis based on right ventricular endomyocardial biopsy is limited because of the segmental nature of the disease, earlier and more severe involvement of the subepicardial than the subendocardial region of the myocardium, and apparent risk in taking biopsy samples from the right ventricular free wall where the histopathologic changes of the disease are typically seen. Asimaki et al.20) recently introduced the immunohistochemical analysis of a conventional endomyocardial-biopsy sample as a new diagnostic test for ARVC/D. They showed an excellent diagnostic value of reduced plakoglobin levels in the biopsy sample, with a sensitivity of 91%, a specificity of 82%, a positive predictive value of 83%, and a negative predictive value of 90%. Importantly, the plakoglobin level was reduced not only in the right ventricle, but also in the left ventricle and interventricular septum. This will allow wider use of endomyocardial biopsy as tissue sampling does not have to be restricted to the histologically abnormal myocardium of the thin-walled right ventricle. Thus, immunohistochemical analysis may facilitate the early and more definitive diagnosis of ARVC/D. However, the utility of this promising test has yet to be proven by a prospective study in a larger cohort.
Magnetic Resonance Imaging Protocol and Findings of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia
With its capability to image the heart without ionizing radiation and high reproducibility, MRI is, at least from a feasibility and safety point of view, best suited for serial assessment of the index cases and screening of asymptomatic family members. The aim of MRI is to assess the morphologic changes, abnormal tissue characteristics, and functional abnormalities of ARVC/D (Table 2 and 3).21-13) The standard MRI protocol includes the sequences for assessment of myocardial thinning, aneurysms, fibrosis, fatty infiltration, and regional and global ventricular function (Table 3). When a patient shows frequent extrasystole during the examination, suppression of ventricular extrasystole with antiarrhythmic medication is important for optimal imaging. Table 4 summarizes the medication used at the Hospital for Sick Children in Toronto. If the patient shows evidence of suppression of ectopy at higher heart rates, either in a 24-hour Holter recording or during an exercise test, our drug of choice is atropine, followed by lidocaine and, in rare cases, isoproterenol. These medications require close observation with continuous monitoring of the heart rate, oxygen saturation, and blood pressure. When medication is used, a physician must be present during the entire examination and prepared to intervene in case of a cardiovascular emergency.
Morphologic changes
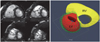
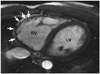
Dilatation of the right ventricle is a nonspecific but common finding of ARVC/D (Fig. 2). In a study by Tandri et al.24) the patients who met the 1994 Task Force criteria for diagnosis of ARVC/D showed a linear correlation between the duration of symptoms and right ventricular end diastolic volume index. Disproportionate dilatation of the right ventricular outflow tract is often seen but is a subjective finding. Although myocardial thinning is considered common pathologically, its recognition at MRI is challenging as the right ventricular myocardium is normally thin. Therefore, it is important to image the pathological areas in strictly perpendicular planes. In adults, myocardial thinning is considered when focal and abrupt reduction in wall thickness to <2 mm is seen.21) Myocardial thinning appears more convincing when it is associated with aneurismal outpouchings. Small multiple outpouchings may give rise to an appearance of focal crinkling, namely an "accordion sign" which becomes more prominent during systole (Fig. 3).23) The "accordion sign" is most commonly seen in the right ventricular outflow tract and subtricuspid regions. Dalal et al.23) showed the "accordion sign" in 60% of the mutation carriers of the family members of the desmosomal mutation-carrying probands of ARVC/D and in none of the individuals without a desomosomal mutation.24) Therefore, the "accordion sign" is considered an early sign of ARVC/D in asymptomatic patients, reflecting an early stage of the disease.23)25) Hypertrophy of the free wall and/or trabeculations of the right ventricle are only occasionally seen (Fig. 2).
Abnormal tissue characteristics
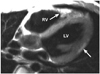
The earlier papers on MRI utilization in the diagnosis of ARVC/D invariably emphasized the ability of MRI to characterize fatty infiltration of the myocardium.26-30) Spin-echo black-blood imaging sequences visualize the fat with high-intensity signals in contrast to intermediate signals of normal myocardium (Fig. 4). Perhaps the ability of the MRI to visualize fat had led to explosive use of MRI in the assessment of ARVC/D. However, identification of myocardial fat was poorly reproducible among readers.31-33) As fibrofatty replacement starts from the subepicardial layer of normal myocardium and extends toward the subendocardial layer, the early histopathological changes are difficult to discriminate from the adjacent epicardial fat. In addition, normal slow flow along the endocardial surface may produce high-intensity signals that may be difficult to differentiate from the signals from fat. Although fat-suppression technique (Fig. 5) can be used for discrimination of the fat from the slow-flow artifact, the detection of myocardial fat remains difficult and subjective. Furthermore, recognition of fat in the normal ventricular myocardium has rendered the diagnostic value of fatty infiltration by MRI of limited value, especially in elderly population and obese individuals who do not have ARVC/D (Fig. 5).34)35) Fatty infiltration with right ventricular thickness ≥ 6 mm, but without regional or global functional abnormalities, is considered distinct from fatty right ventricle in ARVC/D.36) Importantly, fat infiltration is seldom the only abnormality of ARVC/D.21)
Late gadolinium enhancement allows recognition of myocardial fibrosis involving the ventricular septum and the left ventricle.23)37)38) Enhancement of the left ventricle is usually easy to identify. However, it is a real practical challenge to identify the enhancing myocardium of the right ventricle because of the thin thickness of the right ventricular free wall and the presence of a layer of epicardial fat that also shows high-intensity signals (Fig. 6). In addition, the free wall of the right ventricle may normally show a higher signal than the other parts of the myocardium when a surface coil is used. Fat suppression may allow better definition of fibrotic myocardium by eliminating the signal from the epicardial fat. The difficulty in assessment of the right ventricle appears also due to timing of routine late gadolinium imaging to late diastolic phase when the ventricular wall is thinnest. This limitation could be overcome by obtaining late gadolinium enhancement imaging in end-systolic phase when the myocardium is thickened (Fig. 7).30) Left ventricular late enhancement is observed in the setting of global right ventricular dysfunction in the classic form of ARVC/D.10) The predilection sites for left ventricular enhancement are the inferolateral wall and inferior wall-septal junction. Localized late enhancement of the left ventricle occurs frequently without concomitant wall motion abnormality, ventricular dilatation, or significant systolic dysfunction.23)
Functional abnormalities
Regional right ventricular dysfunction is present in most of the patients with ARVC/D (Fig. 8).21)38)40) It is the most sensitive and specific finding in ARVC/D. It represents regional loss of myocytes and precedes changes in global right ventricular function.21) Regional dysfunction is seen in a form of akinesia or dyskinesia, which is often difficult to perceive. The wall motion should be assessed region by region. We find placement of a reference cursor on the center of the ventricular cavity during cine display of the images helpful. Akinetic or dyskinetic motion of the affected wall can be perceived as absence of motion toward the cursor or apparent outward protrusion during systole. Dyssynchronous contraction of the ventricular wall has also been described but is subjective and appears more difficult to recognize.
The modified Task Force criteria of 2010 included akinesia, dyskinesia, or dyssynchronous contraction of the right ventricle as the major criterion when it is associated with overt dilatation or systolic dysfunction of the right ventricle, and as the minor criteria when it is associated with borderline dilatation and systolic dysfunction of the right ventricle.19) ARVC/D is also associated with regional left ventricular dysfunction. Jain et al.41) demonstrated decreased regional left ventricular contraction by using tagged MRI in patients with a definite or probable diagnosis of ARVC/D.
The right ventricle shows progressive reduction in systolic function and dilatation with time (Fig. 8). End stage ARVC/D is characterized by right heart failure with a grossly dilated right ventricle. On the other hand, overtly reduced ejection fraction of the right ventricle is seen only occasionally in early stage of the disease. Global left ventricular dysfunction is seen only in severe cases, although regional dysfunction is not uncommon.41)
Summary of magnetic resonance imaging findings of arrhythmogenic right ventricular cardiomyopathy/dysplasia
Among the described MRI findings of ARVC/D, regional wall motion abnormalities are considered as the most reliable finding. Crinkling of the right ventricular wall with small multiple outpouchings, namely an "accordion sign" appears to be a sensitive morphological finding.23) Quantification of the ventricular volumes provides additional objective data. Late gadolinium enhancement appears to play an important role in detection of left ventricular involvement. Originally regarded as a key MRI finding in ARVC/D, intramyocardial fat is far from reliable, particularly when it is an isolated finding.
Tailored Magnetic Resonance Imaging Approach to Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia in Children
Despite the wide use of MRI as a diagnostic tool in ARVC/D in the last two decades, it has received a more guarded reception than it originally had owing to interobserver variability and lack of standard protocols.17)38) Although MRI appears to be a highly sensitive test in patients fulfilling 1994 Task Force criteria, the diagnosis of ARVC/D can be made without MRI as other imaging modalities are likely to provide the relevant information.38) The most recent report of the North American Multidisciplinary Study of ARVC/D showed a disappointingly low diagnostic performance of MRI compared with other diagnostic tests, including echocardiography and angiography.17) The diagnostic yield of MRI for ARVC/D is significantly lower in the pediatric age group than in adults.43) In fact, it needs to be determined whether MRI is a cost-effective test for the diagnosis of ARVC/D in children and whether the MRI protocol and criteria used for adults are equally applicable to children.
At the Hospital for Sick Children in Toronto, 100 MRI examinations were performed for children with a known or suspected diagnosis or family history of ARVC/D in 2008 using the study protocol listed in Table 3. One study failed because of sustained arrhythmias. Thirty-four individuals showed one or more positive findings (Table 5). Right ventricular dilatation, reduced right ventricular ejection fraction, and regional wall motion abnormalities with thinning of the myocardium or focal aneurysm were the most common findings that were seen in 14-20% of the cases. However, no case showed myocardial fat or positive results of late gadolinium enhancement. In Fogel et al's study,43) only 2 of 81 children referred for MRI evaluation of ARVC/D showed evidence of fatty infiltration in very small areas of the right ventricle. The results of Fogel et al.43) and our study are in contrast to the results of Aviram et al.44) that demonstrated MRI signals compatible with fat in 70% of 26 patients aged 4-17 years. Considering that detection of intramyocardial fat is hampered by a high interobserver variability32) and fatty replacement of the myocardium is the latest pathological manifestation of loss of myocardium, the high incidence of intramyocardial fat in their pediatric population seems questionable. In our routine MRI protocol (Table 3), approximately one third of the scan time is used for imaging the fat and late gadolinium enhancement. On the other hand, positive results for myocardial fat and late gadolinium enhancement are almost exclusively seen in association with other findings, such as ventricular wall motion abnormalities, aneurismal outpouchings, and significant ventricular dilatation. Therefore, the likelihood of positive results for myocardial fat and fibrosis is almost none if cine imaging does not show any abnormality.
As young pediatric patients are less compliant than adults, it is important to perform the sequences with a higher chance of positive finding in the early part of the examination before the patient gets tired and noncompliant. In this regard, cine imaging of the heart in axial, short-axis, and right ventricular outflow tract planes should be performed first, while the fast-spin echo imaging and late gadolinium enhancement imaging are reserved for those who show abnormalities at cine imaging. When the patient has had or shows arrhythmias, an intravenous route is secured for antiarrhythmic medication to ensure proper electrocardiographic gating. In those patients with an intravenous route available, late gadolinium enhancement study can be considered.
In summary, an MRI protocol should be tailored according to the patient's age and likelihood of compliance, as well as the presence of other findings, instead of using the protocol that is used for adults. Importantly, MRI should be performed and interpreted by experienced examiners in these cases to reduce the number of false positive and false negative readings.







 XML Download
XML Download