

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
LEOPARD syndrome (LS) is a rare, multi-systemic, congenital disorder. This disorder is known to be inherited in an autosomal dominant pattern. A missense mutation in the protein-tyrosine phosphatase nonreceptor type 11 (PTPN11) gene is the key mutation responsible for the development of LS.1)2) The diagnosis of this syndrome is based on multiple lentigines with ≥3 cardinal features.3) When lentigines are absent,≥3 other features in patients with LS are diagnostic. In children or young patients without lentigines, LS is diagnosed in the presence of ≥3 of the following criteria: cardiovascular abnormalities, café au lait spots, sensorineural deafness, hyptonia or delayed motor development and distinct facial abnormalities such as hypertelorism, downslanting palpebral fissure, palpebral ptosis and ear abnormalities.2)
The characteristic cardiovascular abnormalities associated with LS are left ventricular hypertrophy (LVH), pulmonary stenosis (PS), electrocardiographic (ECG) abnormalities and coronary artery dilatation.4) All of these abnormalities may progress with age or be detected later in life than other clinical findings.5) Therapeutic plans for cardiovascular abnormalities in LS should be aimed at adverse cardiac events secondary to valvular heart disease or LVH. Here we demonstrate a case of LS who had multiple cardiovascular problems and received successful surgical valve replacement and medical treatment.
Case
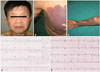
A 47-year-old Korean man presented with palpitations and exertional dyspnea (New York Heart Association class III). On physical examination, he was of notably short stature. He displayed ocular hypertelorism on his face (Fig. 1A) and dermographic lentiginous skin lesions distributed on his right arm and chest (Fig. 1B and C). His height was 134 cm and weight was 36 kg. In cardiac auscultation, a grade 3/6 systolic ejection murmur was heard at the left second intercostal space. He had no family members with whom we could make contact. He showed no communication disability or signs of mental retardation.
An initial 12 lead ECG revealed a sustained ventricular tachycardia (VT) that originated from the apical inferoseptal LV wall (Fig. 1D). Blood pressure and heart rate were 90/60 mmHg and 185/min, respectively. He was immediately given 150 mg of intravenous amiodarone for 10 minutes and the ECG recovered from VT and demonstrated normal sinus rhythm with right bundle branch block pattern (Fig. 1E). An initial echocardiogram showed diffuse LVH (Fig. 2A). Continuous wave (CW) Doppler revealed a severe form of PS {maximal velocity: 4.2 m/s, pulmonic valve (PV) peak pressure gradient: 69.6 mmHg} (Fig. 2B). In addition, the measurement of right ventricular (RV) systolic pressure revealed severe PS (maximal velocity: 4.1 m/s, right atrial pressure: 15 mmHg, RV systolic pressure: 81.9 mmHg) (Fig. 2C).
He had ≥3 characteristic features of LS consistent with diagnostic criteria for LS. We planned to perform PV replacement surgery to relieve heart failure symptoms. Because he responded well to amiodarone, we maintained him on oral amiodarone therapy to suppress the VT. On the evaluation of coronary artery abnormalities by cardiac 64-slice multidetector computed tomography (64-MDCT, Aquilion 64; Toshiba Medical Systems, Tokyo, Japan), the coronary arteries were normal but the pulmonary trunk was significantly dilated (Fig. 2D).
We performed surgical PV replacement with infundibulectomy and transannular RV outflow tract widening. The native PV was replaced by use of a 23 mm Carpentier-Edwards pericardial prosthetic valve. After successful PV replacement, clinical symptoms were significantly improved without any postoperative complications. Nine months later, follow-up echocardiography showed a well-functioning prosthetic PV (maximal velocity: 1.6 m/s, PV peak pressure gradient: 10.1 mmHg) (Fig. 3A) and improved PS (maximal velocity: 2.65 m/s, right atrial pressure: 10 mmHg, RV systolic pressure: 38.6 mmHg) (Fig. 3B). We did a follow-up cardiac 64-MDCT scan to evaluate changes in post-stenotic dilatation of the pulmonary trunk after surgery. Pulmonary trunk size had decreased from 45.4 mm to 37.6 mm after PV replacement (Fig. 3C). There has been no recurrence of heart failure symptoms or VT during the past 12 months.
Discussion
Cardiac involvement in patients with LS is usually characterized by LVH, PS and ECG abnormalities. As the cardiac involvement in LS resembles Noonan syndrome, the differential diagnosis should be made by careful examination.6) In the largest published study on LS, LVH was shown to be the most common cardiac abnormality.4) Asymmetric LVH is more common than apical or concentric hypertrophy. RV hypertrophy may also accompany LVH and PS in about 30% of patients with LS.7) Previous studies have reported that the appearance and degree of LVH occurs concomitantly with the first manifestation of multiple lentigenes.8)
PS is less frequent than LVH in patients with LS. It can be observed in about 40% of reported cases.5) Valvular or infundibular PS is a common type of PS in patients with LS. Mitral or aortic valvular anomalies may occur sporadically as well.
ECG abnormalities occur in about 75% of LS patients, including LVH or biventricular hypertrophy in 46% of them, often in association with q waves, a prolonged QTc, and repolarization abnormalities.4) Arrhythmogenic properties of LVH may induce life-threatening VT or other tachyarrhythmic events in this syndrome. However, the incidence of sudden cardiac death due to ventricular arrhythmia is unclear.
Coronary artery dilatation or aneurysm can be detected incidentally by coronary angiography or cardiac 64-MDCT. However, the clinical significance of coronary artery abnormalities needs further investigation.
Overall prognosis for LS is primarily determined by the presence and severity of cardiac abnormalities. Therefore, proper surgical or medical management should be emphasized. Long-term prognosis of LS seems benign with only mild cardiac involvement. As in our case, the patient with LVH and PS may present with significant symptoms and arrhythmic events during follow-up. Several studies suggest that pathologic and clinical findings may be similar in familial hypertrophic cardiomyopathy and LVH associated with LS. Because our patient showed a favorable response to oral amiodarone therapy, we did not insert an implantable cardioverter defibrillator (ICD) which is recommended for patients with familial hypertrophic cardiomyopathy. Also, the patient showed no premature ventricular complex or non-sustained VT on Holter monitoring during oral amiodarone maintenance. If LVH or arrhythmic events are aggravated on follow-up monitoring, we would consider ICD implantation for primary prevention against sudden death, even if the patient were asymptomatic.
We experienced a case of LS who had multiple cardiovascular abnormalities including PS and LVH, and received successful surgical and medical treatment. Our clinical report suggests that complete cardiovascular screening in patients with LS may be essential and beneficial to determine ideal medical and surgical therapeutic strategies.


 XML Download
XML Download