

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Heart disease is the leading cause of morbidity and mortality in the developed world. Apoptosis, a highly regulated cell death process, plays an important role in numerous pathologic conditions involving the heart,1)2) and the inhibition of apoptosis is emerging as a potential therapeutic strategy. This review provides an overview of the evidence for apoptosis in cardiovascular disease, discusses the molecular pathways that may be involved, and reviews the clinical implications.
Apoptosis in Cardiovascular Diseases
Apoptosis has been shown to be involved in both acute and chronic loss of cardiomyocytes in myocardial infarction, ischemic heart disease, reperfusion injury, various forms of cardiomyopathy, and the development of both acute and chronic heart failure.3-5) Animal and human studies have demonstrated that apoptosis is present in the border zone of the infarcted myocardium in the early phase, confirming the important role of apoptosis in acute myocardial loss after myocardial infarction.6)
Further studies showing that apoptosis is also present months later suggest that apoptosis may also play a role in remodeling and in the subsequent development of heart failure.7) Since cardiomyocyte loss is the most important determinant of morbidity and mortality after myocardial infarction, preventing cardiomyocyte loss becomes a critical issue in the management of myocardial infarction. A better understanding of the regulatory mechanisms of apoptotic signaling is crucial in devising such strategies.
In contrast to acute myocardial injury, the pathogenesis of chronic heart failure is characterized by the progressive loss of cardiomyocytes evolving over months to years. Numerous studies involving human and animal models of heart failure suggest that apoptosis may be an important contributor to cardiomyocyte loss in the setting of heart failure.3) However, since the prevalence of apoptosis is very low in most forms of chronic heart failure (usually <0.1% terminal deoxynucleotidyl transferase dUTP nick end labeling-positive cells), whether or not apoptosis significantly contributes to the pathogenesis of heart failure or is an epiphenomenon associated with end-stage heart failure is still debated.5) Nevertheless, even at a very low level, the contribution of apoptosis over months to years is likely to prove clinically significant in patients with chronic heart failure. The current dilemmas are what forms of cell death (apoptotic vs. non-apoptotic) predominate in chronic heart disease, and whether or not the inhibition of cell death in the form of chronic inhibition therapy will prove beneficial in blocking the progression of clinical heart failure.
Mechanism of Apoptosis
Over the last two decades, much work has been done to identify and elucidate the molecular mechanisms that regulate and execute apoptosis. These studies have shown that apoptosis is a tightly regulated, cell death process that involves close interactions among various pro- and anti-apoptotic molecules. It is generally agreed that apoptosis cannot be strictly identified by only one or two characteristics. In fact, a number of different types of mechanisms have been identified and characterized, such as intrinsic vs. extrinsic pathway or caspase-dependent vs. caspase-independent apoptosis.
Caspase-mediated apoptosis
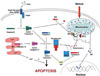
The caspases are a family of cysteine proteases that cleave target proteins at specific aspartate residues.8)9) The caspases are produced as zymogens that are activated after cleavage of their prodomains.8) Caspases are grouped based on structure and function. Initiator caspases possess a long prodomain with a functionally important interacting domain. Caspase-9 and -8 are examples of initiator caspases, which act upstream to initiate and regulate apoptosis, and downstream to activate effector caspases. In comparison, effector caspases, such as caspase-3, are characterized by short prodomains, and generally depend on initiator caspases for activation. Studies show that homologous deletions of specific caspases most often cause tissue-specific or stimulus-dependent effects, rather than a global suppression of cell death.10) These findings suggest that distinct sets of caspases may be involved in specific apoptotic pathways, and they likely act in a tissue-specific manner. In general, caspase-mediated apoptosis occurs either by extrinsic (involving death receptors) or intrinsic (mitochondria-mediated) pathways (Fig. 1).11-16) These two pathways usually converge on a common effector caspase, such as caspase-3, to execute the final morphologic and biochemical alterations that are characteristic of apoptosis.8)
Extrinsic death receptor apoptotic pathway in cardiovascular disease
The death receptor-mediated pathway is initiated by the binding of a death ligand {e.g., Fas ligand (FasL) or tumor necrosis factor-alpha (TNF-α)} to a membrane-bound death receptor (e.g., Fas or TNF-α receptor).14) This interaction leads to the recruitment of a death domain {e.g., Fas-associated death domain (FADD)}, which activates caspase-8 followed by the downstream effector caspases. Several studies suggest that the extrinsic apoptotic pathway has an important pathophysiologic role in the pathogenesis of heart failure.17-23) The importance of FADD, for example, has been demonstrated by gene knockout, which causes embryonic lethality resulting from heart failure and abdominal hemorrhage.17) Components of the death receptor-mediated apoptotic pathway are up-regulated in cardiomyocytes during myocarditis, particularly in immune-mediated cardiomyopathy.18) In human immunodeficiency virus cardiomyopathy, both death receptor- and mitochondria-mediated apoptosis pathways are involved in the apoptosis of cardiomyocytes.19) In addition, several studies have reported that failing human myocardium expresses high levels of TNF-α,20)21) and transgenic mice overexpressing cardiac-specific TNF-α develop dilated cardiomyopathy.22)23) These data suggest that increased levels of TNF-α are detrimental to the heart by activation of the death receptor pathway.
The Fas pathway may be an important mediator of cardiomyocyte apoptosis during ischemia/reperfusion (I/R) in vivo. Various knockouts of the death receptor pathway have been shown to improve cardiac function after I/R injury by inhibiting apoptosis. Mice lacking Fas exhibit reduction in infarct size after I/R.24) However, the heart has high levels of death receptor pathway inhibitors, such as apoptosis repressor with caspase recruitment domain (ARC) and FLICE-inhibitory protein. Indeed, cardiac-specific overexpression of FasL does not cause increased cardiomyocyte apoptosis,23)25) and TNF receptor 1 or 2 knockout in mice does not affect infarct size after coronary artery ligation.26) These findings suggest that although the death receptor-mediated pathway could be important in certain situations (e.g., autoimmune-mediated heart failure), the role of the death receptor-mediated pathway in myocardial infarction or I/R is unclear.
Intrinsic mitochondria-mediated apoptotic pathway in cardiovascular diseases
Mitochondria constitute approximately 30% of the cell volume in cardiomyocytes, and play an essential role by generating adenosine triphosphate (ATP) for cellular function. However, upon apoptotic stimulation, such as oxidative stress and serum deprivation, mitochondria become a critical organelle in the initiation of cell death. In the mitochondria-mediated or intrinsic apoptotic pathway, an apoptotic insult induces the mitochondria to release cytochrome c into the cytosol.11) There mitochondria forms an activation complex, the apoptosome, with apoptotic protein activating factor-1 (Apaf-1) and caspase-9.12)27) Apoptosome formation results in the autoprocessing of caspase-9, as well as the activation of downstream caspases, such as caspase-3.12)15)
The regulation of the release of apoptotic factors, such as cytochrome c from mitochondria, is modulated by the Bcl-2 family of proteins. The Bcl-2 family of proteins can be categorized as either anti-apoptotic (e.g., Bcl-2 and Bcl-xL) or pro-apoptotic (e.g., Bad, Bak, and Bax).28)29) One of the pro-apoptotic members, Bcl-2-interacting protein (Bid), may regulate the interaction between death receptor and mitochondrial pathways. Bid is usually located in the cytosol, but when it is cleaved to 'truncated Bid' (tBid) by activated caspase-8, Bid translocates to the mitochondria and regulates cytochrome c release.30) The protective role of anti-apoptotic Bcl-2 in the heart is demonstrated by the fact that cardiac-specific overexpression of Bcl-2 significantly reduces infarct size after I/R.31)32) Deleting pro-apoptotic Bax also results in reduced infarct size and improved function after experimental myocardial infarction.33)
Additional regulatory mechanisms of caspase-mediated pathways involve caspase inhibitors. Inhibitor of apoptosis proteins (IAPs) are prototypical inhibitors of caspases that block caspase function, usually by directly binding to the caspases.34) Cardiac-specific overexpression of cIAP2 reduces infarct size after I/R in isolated perfused hearts.35) Another important caspase inhibitor is ARC, which is found in high levels in skeletal muscle and heart.36)37) ARC interacts with upstream caspases and has been shown to block caspase-2 and -8, as well as cytochrome c release. The overexpression of ARC decreases infarct size after I/R.38) We have also identified other anti-apoptotic factors, such as HS-1 associated protein-1 (HAX-1), which acts by directly interacting with pro-caspase-9, and prevents its activation.39)
Caspase-independent apoptosis (Fig. 1)
Although caspase activation is most likely the predominant mechanism in the induction of apoptosis, accumulating evidence demonstrates that apoptosis may also be mediated by mechanisms that do not involve caspases.40-42) The so-called caspase-independent pathways involve the release of apoptotic factors, such as apoptosis inducing factor (AIF), from mitochondria to the cytosol followed by translocation to the nucleus, where they cause deoxyribonucleic acid (DNA) fragmentation without concurrent caspase activation.43)44) In contrast to caspase-mediated apoptosis, which is characterized by an oligonucleosomal DNA fragmentation in multiples of -200 bp with an advanced chromatin condensation pattern, caspase-independent apoptosis is characterized by large scale DNA fragmentation (-50 kbp) with an early chromatin condensation pattern.41)42) The potential importance of caspase-independent pathways in the heart is highlighted by the fact that cardiomyocytes contain high levels of endogenous caspase inhibitors, thereby making them relatively resistant to caspase-dependent apoptosis.40) Thus, the role of caspase-independent apoptosis may be amplified in the heart.
Apoptosis-inducing factor
The best documented example of caspase-independent apoptosis involves AIF,41-43)45)46) AIF is a flavoprotein localized in the mitochondrial intermembrane space and is required for oxidative phosphorylation.47) Upon apoptotic stimulation, AIF is released into the cytosol and translocates into the nucleus to induce DNA fragmentation without caspase activation.41) This notion is supported by the fact that microinjection of AIF into cells induces apoptotic changes, such as chromatin condensation, that are not blocked by a caspase inhibitor (zVAD.fmk),41)48) In the heart, AIF has been implicated in apoptosis induced by oxidative stress, ischemia reperfusion, and heart failure in vitro and in vivo.49)50) AIF also accumulates in the cytosolic and nuclear fractions of the heart following I/R.51) We have demonstrated significant activation of caspase-independent apoptosis in the Dahl salt-sensitive rat model of heart failure.52) We have also recently shown that AIF-induced apoptosis is activated in cardiomyocytes, especially in hypertrophic cardiomyocytes.53)
Despite its pro-apoptotic function, AIF has also been shown to possess an essential pro-survival function. Homozygous AIF knockout in a mouse is lethal to embryos,54) and the Harlequin (Hq) mouse, which expresses 10-20% of normal AIF levels, is prone to increased damage from I/R injury.55) In addition, a mouse with cardiac and skeletal muscle-specific knockout of AIF develops severe dilated cardiomyopathy and skeletal atrophy accompanied by defective mitochondrial respiratory activity.56) How, then, is AIF able to function as both a survival and a death-inducing factor? An elegant study by Cheung et al.57) using gene-targeted mice with various AIF mutants demonstrated that AIF is required for cell survival and normal mitochondrial respiration in neurons. On the other hand, during apoptotic stimulation, the pro-apoptotic function of AIF is recognized when AIF is released from mitochondria and translocates to the nucleus, where it promotes DNA damage.
Other factors involved in caspase-independent apoptosis
Other caspase-independent apoptotic effectors have been demonstrated, including endonuclease G (Endo G), serine protease high temperature requirement protein A2 (HtrA2/Omi), and Bnip3. Endo G, a conserved nuclease, is involved in mitochondrial DNA replication with important roles in recombination and repair. Similar to AIF, Endo G translocates from the mitochondria to the nucleus during apoptosis and induces DNA fragmentation independent of caspases.58-60) Endo G and truncated AIF become the essential mediators of apoptosis in a caspase-independent manner in cardiomyocytes.61) Interestingly, Endo G null mice, however, do not have any obvious defects in development or in the regulation of apoptosis.58)59) HtrA2/Omi, a mitochondrial serine protease with pro-apoptotic properties, may also contribute to caspase-independent apoptosis.62) There is evidence that HtrA2/Omi also translocates from the mitochondria to the cytosol during I/R to induce apoptosis. In heart, a specific HtrA2/Omi inhibitor, ucf-101, has also been shown to attenuate apoptosis and decrease infarct size.63)
Other types of cell death
Endoplasmic reticulum-stress death pathway
The endoplasmic reticulum (ER) is responsible for the synthesis and folding of secreted proteins, as well as Ca2+ storage. Several studies have demonstrated a role for ER stress in the pathogenesis of diabetes and heart failure.64) Consistent with these observations, defective ER quality control in transgenic mice with mutant KDEL receptor (a receptor for ER chaperones) causes dilated cardiomyopathy,65) suggesting that apoptosis mediated by ER stress may be a significant contributor to cardiovascular disease. ER stress-induced cell death may occur via two different mechanisms. Under ER stress, activated caspase-12 activates caspase-3, leading to apoptosis.66) The second death-signaling pathway activated by ER stress is activation of a transcriptional program via up-regulation of the transcription factor, CHOP/GADD 153. CHOP activates the transcription of genes encoding pro-apoptotic proteins, including the BH3-only protein, Puma.67) Recently, it has been suggested that Puma is a critical component of ER stress-induced apoptosis in cardiac myocytes.68) The Bcl-2 proteins have been shown to localize to the ER, where they can regulate the levels of Ca2+ stored in the ER.69)
Non-apoptotic cell death
This review is focused on apoptotic cell death, but non-apoptotic mechanisms, such as necrosis and autophagy, are also important cell death processes in heart. Necrosis, which has often been viewed as an accidental and uncontrolled cell death process, might also be a highly orchestrated type of programmed cell death, such as apoptosis, and this subset of regulated necrosis is termed necroptosis.70) Unlike apoptosis or necrosis, autophagy is primarily involved in survival. Autophagy enables cells to dispose of cytoplasm and organelles by fusing vesicles containing cellular components and lysosomes.71) However, several studies have demonstrated that autophagy has features resembling apoptosis, including a possible association with the caspases and Bcl-2.72-75) We also recognize that there is considerable controversy at present around differentiating the various types of cell death. There may be a spectrum of different mechanisms, and which mode of cell death predominates depends on the specific type of apoptotic stimuli, the degree of insult, and the intracellular ATP concentration. These are important and controversial issues at this time, and further studies are needed to clarify these modes of cell death.
Inhibition of Apoptosis as Therapy for Cardiovascular Disease
Since apoptosis is implicated in the pathogenesis of many different cardiovascular diseases, the inhibition of apoptosis promises to be an extraordinarily important target for therapeutic intervention. Even though the therapeutic targeting of apoptotic pathways has potential in the treatment of heart failure, several important questions remain to be answered. First, it has not been shown whether or not the inhibition of apoptosis can delay or prevent the development of heart failure. It is possible that inhibiting apoptosis may simply result in the activation of another mode of cell death, such as necrosis, which may have more deleterious effects on neighboring cells and ultimately a worse outcome. Although the early studies on animal models of heart failure have been encouraging, the long-term consequences of inhibiting apoptosis in the heart are not known. Second, the safety of anti-apoptotic therapy has not been rigorously tested. Apoptosis is needed for the normal functioning of various cell systems, such as the immune system, and an excessive inhibition of apoptosis is associated with lymphoma or autoimmune disorders. Therefore, the chronic systemic inhibition of apoptosis may have significant deleterious consequences in non-cardiac organs. Third, anti-apoptotic therapy for heart failure may not apply to all types of heart failure. The most ideal conditions for anti-apoptotic intervention, in our opinion, occur in transient and acute insults, such as reperfusion. During reperfusion, cardiomyocyte apoptosis occurs at a high rate during a defined time period; thus, a short treatment period may be highly effective. Moreover, a short therapeutic course has the additional benefit of minimizing the possible deleterious side effects arising in other organ systems.
Future Outlook and Conclusion
It is clear that apoptosis plays a critical role in the pathogenesis of various cardiovascular diseases and the inhibition of apoptosis promises to be an extraordinarily important target for therapeutic intervention. However, more work is necessary to understand the molecular mechanisms that govern these processes, and the significance of apoptosis in heart failure. For example, although caspase inhibition has been shown to reduce the acute loss of myocardium in various animal models,76)77) caspase inhibition might not be completely effective in blocking apoptotic cell death.78) With the potentially significant contribution of caspase-independent apoptotic cell death in the heart, it is important to better define the role of the caspase-independent pathway in cardiac apoptosis at this time. Further work must be carried out in well-defined experimental frameworks that are tissue-targeted and time-specific, with clear quantitative end points. Only then will we be able to conduct meaningful human studies to answer the question of whether the inhibition of apoptosis in heart failure will translate into clinical benefit.
 XML Download
XML Download