

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
In 1992, Brugada syndrome (BS) was first described as a new clinical entity characterized by a typical electrocardiogram (ECG) pattern {right bundle branch block (RBBB) and persistent ST-segment elevation in right precordial leads} and sudden cardiac death.1)2) After the first formal description in the medical literature, this clinical entity has been increasingly diagnosed.2-4) In patients with BS, sudden onset of ventricular tachycardia may develop; when this arrhythmia persists, it can degenerate into ventricular fibrillation, resulting in sudden cardiac death. BS is known as a disease that is inherited via an autosomal dominant mode of transmission with varying penetration.2)5) Although this syndrome is commonly known to manifest in adulthood, usually in the fifth decade, some pediatric cases have been reported, including one of a 2 day-old infant.6)
To the best of our knowledge, there have been no reports on BS in Korean children. We report on childhood BS in two families.
Cases
Case 1
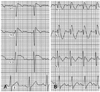
A one-month-old male infant presented to the outpatient-department (OPD) for systolic murmur. Echocardiogram revealed perimembranous ventricular septal defect (VSD) with septal aneurysm. ECG was obtained and showed right axis deviation with possible RBBB (Fig. 1A). The patient visited OPD regularly, and no remarkable symptoms were found. Because the VSD was of moderate size without significant hemodynamic compromise, the patient underwent routine observation for VSD. When he was 4 months old, ECG revealed significant QRS widening in right precordial leads, with saddle back ST elevation in the V3 lead (Fig. 1B). The pattern of ECG changed progressively until 6 months of age, when definitive ST elevation with T wave inversion of Brugada type 1 pattern was observed (Fig. 1C). At 24-hour Holter monitoring, rare, premature ventricular contractions (<1%) were recorded without run. The patient had no family history of syncope or sudden death. Initial ECGs of family members, including his parents and sister, were negative. However, ECG taken during the 2 mg/kg flecainide challenge test revealed a type 1 Brugada pattern in both father and mother (Fig. 2). A familial genetic sequencing study for SCN5A was performed. Deoxyribonucleic acid (DNA) sequencing analysis of the cardiac sodium channel (SCN5A) revealed a G to T base substitution at nucleotide position 4035 on exon 23 that led to replacement of tryptophan by cysteine at codon 1345 (c.4035G>T, p.W1345C, heterozygote) in domain III of the sodium channel (Fig. 3B). Study of the patient's father revealed no mutation, but the same novel mutation as that of the patient was found in a study of the mother (Fig. 3C). No other SCN5A mutations were found. This mutation was not detected in 101 unrelated control subjects. Genetic studies on maternal family members are recommended. This child with manifest Brugada type ECG with SCN5A mutation is asymptomatic at this point; thus, he is being closely monitored without treatment.
Case 2
A 12-year-old boy and his 10-year-old sister were referred to the pediatric cardiology OPD for familial history of sudden death. Their father had died of sudden cardiac arrest after some alcohol intake at the age of 44 years. He had no history of previous syncopal attack and no other significant medical history. Cardiac arrest was suspected to be due to ventricular tachycardia associated with BS by the attending physician. The patients' paternal great grandfather died during sleep in his fifth decade. The brother and sister had no medical history or symptoms, such as chest discomfort, syncope, or palpitation. Their ECGs and Holter monitorings revealed no significant abnormality. Echocardiogram showed normal cardiac structures. On arrhythmia drug test with flecainide, the boy showed typical coved type ST elevation in the precordial leads V1 to V3 (after 13 doses of flecainide 8 mg, 2.7 mg/kg) (Fig. 4A), and the girl also revealed typical coved type ST elevation in the precordial leads V1 to V3 (after 10 doses of flecainide 10 mg, 1.9 mg/kg) (Fig. 4B). Programmed ventricular stimulation test showed no inducible ventricular tachycardia in either patient. Genetic study for SCN5A was performed for the female patient, but no causative mutation was detected. Both patients are in good condition, without medication or symptoms after discharge. Implantable cardioverter-defibrillators (ICDs) are under consideration if symptoms or signs develop.
Discussion
BS is a disease of adulthood, typically resulting in sudden death during the forties6)7); however, some cases have reported on childhood BS in patients as young as a few days old. Since it was first formally reported in 1992, numerous studies and reports on the clinical characteristics or the genetic and molecular aspects of the disease have appeared.2)8)9) Now BS is regarded as responsible for 4% to 20% of all sudden death, and up to 20% of sudden death without structural heart disease.6)9) Characteristic ECG patterns were identified soon after the first report: 1) type-1 ECG pattern was described in the initial report, where a coved ST-segment elevation greater than or equal to 2 mm is followed by a negative T wave in more than 1 right precordial lead (V1-V3); 2) type-2 ECG pattern is characterized by an ST-segment elevation, followed by a positive or biphasic T wave that together compose a saddle-back morphology; 3) type-3 ECG pattern is characterized by a right precordial ST-segment elevation less than or equal to 1 mm with either a coved-type or a saddle-back morphology.2)10) Although all of these 3 ECG patterns can be identified in patients with BS, only the type-1 ECG pattern now defines diagnosis of the syndrome.6)10) The proband of the first family in this report showed dramatic progression of the Brugada type ECG pattern from simple RBBB to type 1 pattern during the first half of the boy's infancy (Fig. 1). Because the patient in case 1 had VSD and septal aneurysm, the initial RBBB change on ECG could possibly be associated with the malformation itself. However, as demonstrated in Fig. 1, ECG progression to typical coved type ST segment elevation in V1-V3, which is the characteristic feature of BS, could have led to the diagnosis of BS. BS can be diagnosed when a type-1 ECG pattern is observed in more than 1 right precordial lead (V1-V3), with or without a sodium channel blocker agent, and in concurrence with one of the following: documented ventricular fibrillation, polymorphic ventricular tachycardia, inducibility of ventricular arrhythmias with programmed electrical stimulation, family history of sudden death before the age of 45 years, presence of coved-type ECG in family members, syncope, and nocturnal agonal aspiration.2)6)10)
Although some sporadic cases of BS have been reported, it is now understood as an inherited disease, in which a familial occurrence was noted in approximately half of patients.2)4) The first description of a related mutation appeared in 1998 and identified the mutation in SCN5A, the gene encoding the alpha subunit of the cardiac sodium channel.11)12) To date, 100 or more other mutations found in SCN5A have been associated with this syndrome, and a second locus was found on chromosome 3p22-25 without identification of the causative gene.2)13) However, only 20-25% of BS shows mutations in SCN5A. Our first proband with infantile manifestation had a novel mutation (c.4035G>T, p.W1345C, heterozygote) equal to that of the mother. However, the mother was asymptomatic, and Brugada type ECG was uncovered only by flecainide injection. This may be due to low penetrance. However, there may be another explanation for the spontaneous change of ECG in case 1 during infancy. This case may have another undefined predisposition because the father of the child showed equivocal type 1 Brugada pattern ST elevation by flecainide, despite having no documented SCN5A mutation. As the SCN5A mutation was not found in up to 70% of BS cases, we can speculate that the father of the patient may have an undefined or unknown mutation causing BS or a genetic polymorphism with predisposal to BS type ST change under certain provocations. On the other hand, we still cannot exclude the possibility of a false positive result for the father on the flecainide provocation test, as the specificity of the provocation test has been reported to be 80% in adults, leaving a 20% false positive rate. However, it should be noted that racial differences could be reflected in the specificity of the provocation test. As a study targeting both adults and children in the Korean population has not yet been reported, further study targeting the Korean population may help to explain the discrepancy between results from genetic analysis and the provocation test. Current opinions do not support the role of genetic analysis in estimation of risk for BS5); however, it may be helpful in detection of asymptomatic carriers of the mutation, who may then be provided with clinical monitoring. SCN5A mutations resulted primarily in the production of either a non-conducting sodium channel or accelerated recovery of the sodium channel. The functional effect of the mutations in transmembrane domain III, W1345C, has not yet been documented.
Several factors have been known to trigger typical Brugada-pattern ECG and polymorphic ventricular tachycardia in susceptible individuals. These factors include autonomic imbalance, electrolyte imbalance, fever, myocardial ischemia, and drugs, including antiarrhythmic agents, psychotropic drugs, alpha adrenergic agonists, potassium channel opening drugs, and first generation antihistamines.5)6)
The only proven treatment option for BS is an ICD.14) Within the current guidelines, implantation of ICD is indicated for patients with BS and aborted cardiac arrest who receive optimal long-term treatment and whose life expectancy in good general condition exceeds one year.5)15)
In the first report of BS, 3 of the 8 patients were children1); however, little information on this syndrome during childhood has been available. It can manifest during childhood, and symptoms may appear during febrile episodes.16) Symptomatic childhood patients, particularly those presenting with a spontaneous type-1 ECG, may be at a high risk for cardiac events during a relatively short period of follow-up.2) The first description of BS in children appeared in the literature in 2000.7)17) Since that time, many cases of pediatric BS have been reported. In three Japanese studies describing the prevalence and clinical course of childhood BS,7)18-20) typical ECG patterns of BS were found less frequently than in adult studies; however, this prevalence increased with age. The nature of BS is known to be dynamic, and this variability is more prominent in children.20)
To the best of our knowledge, there have been no reports on BS in Korean children. We reported on childhood BS in two families; one family had a novel SCN5A mutation. Physicians should be aware of this potentially lethal disease, which may manifest even in infancy, resulting in sudden infant death syndrome.4)17)



 XML Download
XML Download