

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Following acute myocardial infarction, re-establishing coronary blood flow with the rapid use of reperfusion strategies such as thrombolysis or primary coronary intervention is essential to salvage viable myocardium. However, paradoxically, the process of reperfusion can itself result in myocyte death, a phenomenon referred to as "lethal reperfusion injury".1) Apoptosis and necrosis are the two major distinct types of cell death of cardiomyocytes that have been associated with ischemia and reperfusion. Particularly, apoptosis-induced myocyte death is thought to play an important role during the early stages of reperfusion.2-4)
The exact mechanisms underlying myocardial reperfusion injury are not known. However, reactive oxygen species (ROS) are formed in excessive amounts within the first few minutes following reperfusion and are considered important to myocardial reperfusion injury.5) In addition, excessive ROS during the early stages of reperfusion increases the activity of mitogen-activated protein (MAP) kinase and induces apoptosis following reperfusion.6-8)
Anti-oxidant treatments have been reported to prevent myocardial reperfusion injury.9)10) Among the anti-oxidants, alpha-lipoic acid (ALA) thiol antioxidant, has been shown to preserve cardiac function during ischemia-reperfusion while acting as a co-factor for mitochondrial dehydrogenase.11-13) Several authors have reported that low-dose ALA reduces myocardial reperfusion injury in vitro.13)14) To date, however, few studies have reported on the effectiveness of ALA in preventing apoptotic myocardial reperfusion injury in vivo.
We examined whether the administration of ALA prior to reperfusion therapy would reduce apoptotic reperfusion injury of myocytes in an ischemia-reperfusion rat model.
Materials and Methods
Experimental animals
Healthy male Sprague-Dawley rats weighed 250-300 g were divided into an ALA treatment group and a control group. The rats in both groups received 45-minutes of ischemia. The infarct size of the myocardium was assessed 45 minutes following reperfusion. Apoptosis staining, ROS synthesis and the activity of MAP kinase were assessed 10 minutes following reperfusion. To confirm the dose of ALA, at which the infarct size was suppressed most effectively, ALA 10, 25, 50 and 100 mg/kg was administered to the right ventricle 10 minutes prior to the reperfusion. The infarct size was smallest in the ALA 25 mg/kg group. Following this, apoptosis ROS synthesis and MAP kinase activity were measured in the ALA 25 mg/kg group.
To induce cardiac ischemia, chloral hydrate (100 mg/kg) (Merck, Germany) was injected intra-abdominally for anesthesia. In the supine position, the rats were fixed onto an experimental plate. An endotracheal tube was placed using a 16-G tube and ventilation provided. The sternum was incised and the heart exposed. The proximal part of the left anterior descending artery was ligated using No. 6 silk and maintained for 45 minutes. Confirmation of myocardial infarction was based on decreased movement of the left ventricle and change of the coloring to blue. After the indicated time, the ligated suture was released. Reperfusion was confirmed by the return to normal coloring of the left ventricle. Thereafter, the rats were sacrificed according to schedule. During all experimental procedures, the temperature of the rats was maintained at 37℃ with a white lamp.
The measurement of the infarct size
To measure the infarct size based on the dose of ALA in all groups including: ALA 10 mg/kg (n=5), 25 mg/kg (n=5), 50 mg/kg (n=5), 100 mg/kg (n=5) and the control group (saline 0.5 mL) (n=5), propidium iodide (PI) 1 mg (Sigma Chemical Co.) was injected into the right ventricle over 30 minutes following reperfusion. PI, 1 mg was diluted in saline 0.5 mL. Forty five minutes following reperfusion, the left anterior descending artery was ligated again. Then, in order to differentiate between the ischemic and normal areas, polystyrene/divinylbenzene fluorescence (Duke Scientific) was injected into the proximal part of the aorta.15) The rats were then sacrificed and stored in a -70℃ refrigerator. Thereafter, the samples were fixated in 4% paraformaldehyde for two hours. Using a vibrotom, the left ventricle was vertically sectioned at a thickness of 400 µm. Following this, the basal, middle and apical areas were selected and the infarct size of myocardium was measured using a polarized microscope. On polarized microscopy, the normal area was observed as a green color, the ischemic area was black and the infarct area was a red color. The infarct size was defined as the ratio of the infarct area relative to the ischemic area of the myocardium.
The measurement of apoptosis
To measure apoptosis at the myocardial infarction site, terminal deoxynucleotidyl transferase mediated biotin-dUTP nick-end labeling (TUNEL) staining was performed in the ALA 25 mg/kg group (n=5) and the control group (n=5), using an in situ apoptosis detection kit (TaKaRa Shuzo Co.). The cardiac tissue was attached to a glass slide and rinsed with 0.01 M phosphate buffered saline (PBS) (pH 7.4), and then treated with proteinase K (20 g/mL) at room temperature for 20 minutes. The sample was again rinsed with PBS. To suppress peroxidase activity, intrinsic peroxidase activity was blocked using 3% H2O2 at room temperature for 5 minutes. Following the rinsing, the sample was treated with a permeability buffer on ice for 2-5 minutes. Fluorescein-dUTP (TdT+labeling safe buffer) was used for the labeling performed at 37℃ in an incubator for 90 minutes. Following rinsing, the anti-FITC HRP conjugate underwent antibody reaction using a 37℃ incubator. Then, the color was developed using DAB (5 mg/mL in Tris buffer, pH 7.0) at room temperature for 10-15 minutes. The degree of apoptosis was then observed using a light microscope. On light microscopy, with the use of a digital camera, the microscopic images were converted to a digital format and then saved in a computer. An automatic system for the imaging analysis was used to measure the number of cells that were positive for TUNEL staining.
The measurement of reactive oxygen species generation
To measure ROS generation, 5-(and-6)-chloromethyl-2',7'-dichlorodihydro-fluorescein diacetate (CM-H2DCFDA) (Molecular Probes Inc., Eugene, USA) 100 µg was directly injected into the right ventricle 10 minutes prior to the ligation of the left anterior descending artery in the ALA 25 mg/kg (n=5) and control groups (n=5). Ten minutes prior to reperfusion, ALA or saline was administered to the right ventricle. Ten minutes following reperfusion, the rats were sacrificed and then stored in a -70℃ refrigerator. Thereafter, with the use of a freezing microtom, the left ventricle was sectioned at a thickness of 40 µm in the vertical direction of the long axis. Following this, basal, middle and apical areas were selected. Then, using a polarized microscope, the fluorescent development of a clear green color was noted, which reflected the ROS.16)17) The amount of ROS synthesis was analyzed using image pro plus in the areas of ischemic injury. The images of the original green-colored samples were converted to an 8 bit gray color. In the areas of ischemic injury, the values of the gray scale were averaged and the expression of ROS defined. The amount of ROS generation that was calculated in the control group was summed. Then, the average was defined as the amount of ROS expression, which was considered 100% compared to the ALA 25 mg/kg group. The amount of ROS generation in the ALA 25 mg/kg group was obtained by calculating the average value of the gray scale in the areas of injury in each experimental animal. This value was calculated as a percentile compared to the values for the control group.
The measurement of mitogen-activated protein kinase activity
Among the MAP kinases, pERK 1/2 and pJNK 1/2 were measured in the rats after they were sacrificed in both the ALA 25 mg/kg (n=5) and the control groups (n=5) 10 minutes following reperfusion. The infarction area of the left ventricle was extracted and then frozen using liquid nitrogen. The sample was stored in a -70℃ refrigerator. To isolate the protein from cardiac muscle, the cardiac muscles were converted to a powder using liquid nitrogen and a grinder. The powder of cardiac muscle was diluted in a histolysis buffer solution (20 mM Tris-HCl pH 8.0, 1% NP-40, 150 mM EDTA, 10% glycerol, 0.1% beta-mercaptoethanol, 0.5 mM DTT) at a ratio of 1 : 5 of weight/volume, and maintained in an ice bath for more than an hour. Centrifugation of the sample was performed at 4℃ at 12,000×g for 15 minutes. Then, the supernatant protein was harvested. Thereafter, using the Bradford method,18) the protein concentration was quantified using a spectrophotometer (Beckman Co., USA).
Protein electrophoresis was performed using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The protein was mixed with a sample buffer (0.125 M Tris pH 6.8, 2% SDS, 25% glycerol, bromophenol blue, 2-mercaptoethanol) at 100℃. The protein content (80 µg), which was mixed with a histolysis buffer solution, underwent electrophoresis with a 12%- and 15%-gradient. The electrophoresis was performed using a mini gel electrophoremeter (SE 600 Hoefer Sci. Ins), at 90 V for two hours. Following electrophoresis, the gel was stained using Coomassie brilliant blue R-250. Destaining was carried out with destaining solution (10% acetic acid and 10% methanol) and the protein bands were confirmed.
With the use of protein transfer equipment (Hoefer Semiphor, Phamacia Bio.), the protein gels were transferred to polyvinylidene difluoride (PVDF) membranes of 0.45 µm in thickness (Millipore Co., USA) for two hours. To block the non-specific binding of primary antibodies, this PVDF membrane was placed in a blocking buffer where Tris buffer saline (TBS) (pH 7.6) was dissolved in a 3% bovine serum albumin and then the reaction was performed for an hour. The primary antibody, pERK 1/2 (1 : 500) (Cell Signaling Technology, USA), was reacted overnight at 4℃ and then rinsed with 0.05% Tween-20-TBS (TBST) three times. Thereafter, the secondary antibody, goat rabbit immunoglobulin G (IgG) conjugated AP (Santa Cruz, USA), was diluted at a ratio of 1 : 2,000 and the reaction was performed for one hour. Following rinsing with TBST, rinsing with alkaline phosphates (0.1 M Tris, 0.1 M NaCl, 0.01 M MgCl2) was performed. Thus, the color was developed with NBT and BCIP. In addition, with the same conditions, as the controlled experiment for pERK 1/2, the inactive form of ERK 1/2 protein expression was observed. The expression of pJNK 1/2 and JNK 1/2 were also observed in the same manner as ERK.
To analyze the degree of the expression of pERK 1/2 and pJNK 1/2, on Western blot analysis, with the use of a scanner (HP4P), the pattern of the proteins' color was developed on the PVDF membrane and then converted to a digital format. Then, the data was saved in a personal computer. With the use of an imaging analysis program (UN-SCAN-IT gel, USA), the area and density of each protein expression were measured. Then, these two parameters were multiplied and the volume of each protein was thereby calculated. The expression of active proteins, pERK 1/2 and pJNK 1/2, were defined based on the final amount of expression compared to the values of ERK 1/2 and JNK 1/2. In addition, the relative ratio of expression was calculated based on the amount of expression of the active forms noted in the sham surgery group (n=2).
Results
The effect of alpha-lipoic acid on the infarct size
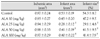
The infarct size based on the dose of ALA was significantly smaller in the ALA 25 mg/kg (29.1±4.8%, p<0.0001), ALA 50 mg/kg (41.5±9.5%, p<0.05), and ALA 100 mg/kg groups (41.4±7.9%, p<0.05) compared to the controls (54.3±8.7%). However, the ALA 10 mg/kg group (47.2±9.5%, p=0.192) did not differ significantly from the control group. The ALA 25 mg/kg treatment group had the smallest infarct size (Fig. 1) (Table 1).
The effect of alpha-lipoic acid on apoptosis
Following TUNEL staining, on light microscopy, apoptotic nucleoli, which were stained as a dark brown color, indicating reperfusion injury due to apoptosis, were frequently detected in the control group. By contrast, this finding was rarely seen in the ALA 25 mg/kg group (Fig. 2).
The effect of alpha-lipoic acid on reactive oxygen species generation
In the ALA 25 mg/kg group, the color development indicating the synthesis of ROS was only observed in 41.6% (p<0.0001) compared to 100% in the control group (Fig. 3).
The effect of alpha-lipoic acid on the activation of mitogen-activated protein kinases
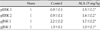
In regard to the pattern of the expression of pERK 1/2 with regard to anti-apoptotic effects and pJNK 1/2 with regard to apoptotic effects, the relative ratio of expression was calculated compared to the sham surgery group in the controls and the ALA 25 mg/kg group (Fig. 4). The expression of pERK 1/2 was significantly increased in the ALA 25 mg/kg group compared to the control group as shown with pERK 1 (0.8±0.1 times vs. 1.5±0.2 times, p<0.05) and pERK 2 (0.9±0.1 times vs. 1.6±0.2 times, p<0.05) (Fig. 5A) (Table 2). In addition, the expression of pJNK 1/2 was significantly decreased in the ALA 25 mg/kg group compared to the control group as shown with pJNK 1 (2.2±0.2 times vs. 1.7±0.2 times, p<0.05) and pJNK 2 (1.3±0.1 times vs. 1.0±0.1 times, p<0.05) (Fig. 5B) (Table 2).
Discussion
In the current study, the administration of ALA prior to reperfusion significantly reduced the infarct size in rats after myocardial ischemia-reperfusion. The administration of ALA 25 mg/kg prior to reperfusion significantly reduced apoptotic cell death in the myocardium. This anti-apoptotic effect of ALA was observed to be closely associated with the inhibition of ROS generation, the increased activity of pERK 1/2 and the decreased activity of pJNK 1/2.
Prompt reperfusion is essential for the survival of cardiac muscle after an ischemic event. In addition to the cardiac injury caused by ischemia, the reperfusion itself can promote cardiac injury and make it worse. This phenomenon is referred to as reperfusion injury.1) In myocardial ischemia-reperfusion, the myocardial injury is caused by necrosis and apoptosis. Kajstura et al.19) and Fliss et al.20) reported that myocardial injury caused by apoptosis was significantly more prevalent than injury due to necrosis in a 2-hour ischemia model and a 45-minute ischemia and a 1-hour reperfusion model. Several investigators have reported that apoptosis was a more critical factor than necrosis with regard to myocardial injury in ischemia-reperfusion animal models, and that apoptosis was significantly increased by reperfusion.2-4)
The exact mechanisms underlying myocardial reperfusion injury is not known. However, ROS are formed in excessive amounts within the first few minutes following reperfusion and are considered a major factor involved in myocardial reperfusion injury.5)21-23) To minimize reperfusion injury, powerful antioxidants have been administered to reduce ROS generation and thereby decrease myocardial injury.17)24)25) Among the antioxidants, ALA, a thiol compound, is a cofactor for mitochondrial dehydration enzymes. In the tissue mitochondria, it is reduced to dithiol dihydrolipoic acid (DHLA), an effective form. DHLA has been reported to play a role in preserving myocardium function by normalization of the intracellular pH, increased mitochondrial ATP synthesis and decreased ATP hydrolysis in the setting of myocardial ischemia-reperfusion.12)13) According to Schonheit et al.13) a low-dose of ALA improved cardiac function following reperfusion in an in vitro study. By contrast, a high-dose of ALA increased cardiac damage following reperfusion. Based on these findings, the effect of ALA treatment on the prevention of reperfusion injury was thought to be dose-dependent. According to Cao et al.14) a minimal dose of ALA increased intrinsic antioxidants and the concentration of phase 2 enzymes with defense against oxidation, in an experiment using cultured cardiac cells from rats. Thus, ALA has been reported to reduce myocardial injury caused by ROS.
ALA has been reported to prevent ischemia-reperfusion injury in a dose-dependent manner in vitro. To date, however, few studies have examined the effects of ALA on the prevention of myocardial ischemia-reperfusion injury in vivo. In the current study, the infarct size of the myocardium was differentially suppressed depending on the dose of ALA administered 10 minutes prior to reperfusion. Compared to the control group, the ALA 25 mg/kg group (p<0.0001) had the smallest infarct size. In addition, the infarct size was also suppressed in the 50 mg/kg (p<0.05) and the 100 mg/kg groups (p<0.05). However, the infarct size was not significantly suppressed in the ALA 10 mg/kg group. Furthermore, in the ALA 25 mg/kg group compared to the control group, apoptosis was significantly suppressed. These results indicate that ALA suppressed apoptosis and reduced myocardial injury during reperfusion.
Little is known about the mechanisms that cause myocardial injury due to apoptosis as a result of ischemiareperfusion in the myocardium. Immediately after reperfusion, however, an excessive degree of ROS generation and the activation of MAP kinase might be involved. ROS generation reached the highest levels within a minute following reperfusion and thereafter persisted. An excessive degree of ROS generation is known to play a crucial role in the pathogenesis of myocardial injury due to apoptosis immediately after reperfusion.21-23) The ROS can be synthesized from endothelial cells, inflammatory cells and cardiac cells through a variety of enzyme activity. Particularly in the early stage of reperfusion, an excessive amount of ROS generation in the endothelial cells and cardiac cells stimulates inflammatory cells and the activated inflammatory cells release ROS. Thus, ROS plays a key role in the early and late stages of apoptosis.22)26)27) ROS generation reached maximum levels within a minute following reperfusion. Therefore, to minimize the reperfusion injury, antioxidant treatment must be performed prior to reperfusion.
Bolli et al.23) assessed myocardial contractility in groups where treatment was performed 15 and 1 minute prior to reperfusion and 1 minute following reperfusion with a control group in a canine myocardial ischemia-reperfusion model. In the groups where the treatment was performed 15 and 1 minute prior to reperfusion, compared to the control group, the myocardial contractility was significantly improved. In the group where the treatment was performed 1 minute following reperfusion, however, there was no significant difference between the treated and control groups. The investigators concluded that treatment must be performed prior to reperfusion to prevent reperfusion injury. In the current study, ALA was administered 10 minutes prior to reperfusion. On polarized light microscopy, using a detector for active oxygen, CM-H2DCFDA, in the ALA 25 mg/kg group compared to the control group, showed that the generation of ROS was significantly suppressed at the site of myocardial infarction (p<0.0001). These findings suggest that the cardioprotective effects of ALA might be closely associated with the suppression of apoptosis due to decreased generation of ROS.
It is not known whether ROS is involved in the activation of the protein kinase pathway, which is associated with the induction of apoptosis following myocardial ischemia and reperfusion. An excessively produced ROS during the early stages of reperfusion activates the inflammatory cells and augments the intracellular calcium levels. In addition, it increases the secretion of pro-apoptotic genes and activates MAP kinase, nuclear factor-κB (NF-κB) and tumor necrosis factor-α (TNF-α), and thereby induces apoptosis. Thus, it is involved in triggering myocardial injury.28) Protein kinase pathways that are activated by ischemiareperfusion of the myocardium include MAP kinase, PI-3 kinase/Akt and tyrosine kinase. MAP kinases can be divided into three types including the extracellular signal-regulated kinase (ERK 1/2), c-Jun-NH2-terminal kinase 1/2 (JNK 1/2) and the p38 α/β kinase.7) MAP kinase belongs to a class of serinethreonine protein kinases that are activated in response to the various stimuli. ERK 1/2 is involved in the survival of cells and JNK 1/2 is involved in apoptosis; thus, it is involved in maintaining the balance between survival and apoptosis.29)30) According to Yue et al.,29) in a myocardial ischemia-reperfusion rat model, the activation of ERK 1/2 reached the highest levels 10 minutes following ischemia and in 10 minutes following reperfusion. When PD98059 selectively blocked the ERK 1/2 pathway, the degree of apoptosis in the myocardium was significantly higher compared to controls.
Thus, it was demonstrated that pERK 1/2 has anti-apoptotic effects in myocardial ischemia-reperfusion. In addition, Ferrandi et al.30) reported that the size of the infarction site was decreased and the apoptosis of cardiac muscles was markedly reduced after activation of the JNK 1/2 pathway was blocked following the use of AS601245, a JNK 1/2 inhibitor, in a myocardial ischemia-reperfusion rat model. Thus, it was demonstrated that pJNK 1/2 was involved in the apoptosis of myocardial ischemia-reperfusion. These findings suggest novel treatment modalities that increase pERK 1/2 involved in cell survival and suppress pJNK 1/2 involved in apoptosis; modulation of these factors might prevent myocardial reperfusion injury due to apoptosis. In the current study, the administration of ALA 25 mg/kg prior to reperfusion resulted in significantly increased pERK 1/2 activity 10 minutes following reperfusion (p<0.05) compared to controls. In addition, the activity of pJNK 1/2 was significantly decreased (p<0.05). These findings suggest that the cardioprotective effects of ALA treatment prior to reperfusion were closely associated with the suppression of apoptosis due to increased pERK 1/2 and decreased pJNK 1/2.
In conclusion, the results of this study show that ALA therapy prior to reperfusion significantly reduced the infarct size in a myocardial ischemiareperfusion rat model. The cardioprotective effects of ALA might be associated with the anti-apoptotic effects of ALA. The anti-apoptotic effects of ALA were closely associated with the inhibition of ROS generation, increased activity of pERK 1/2 and decreased activity of pJNK 1/2. The cardioprotective effects of ALA with regard to reperfusion injury in vivo suggest that ALA therapy might be a novel treatment modality for minimizing reperfusion injury in patients with acute myocardial infarction in the clinical setting. Additional studies are needed to examine the effects of ALA administration in a large series of patients with acute myocardial infarction.





 XML Download
XML Download