

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Fabry disease is a rare X-linked disorder caused by mutations in the GLA gene encoding the lysosomal enzyme α-galactosidase A (α-Gal A). It results in a failure to catabolize lipids containing α-D-galactosyl moieties and subsequent progressive intracellular accumulation of glycosphingolipids in various tissues, including skin, kidneys, vascular endothelium, ganglion cells of the peripheral nervous system, and heart. The disorder is caused by mutations in the α-Gal A gene located in the X chromosomal region Xq22.1) Cardiac involvement is characterized by progressive left ventricular hypertrophy (LVH) that mimics the morphologic and clinical features of hypertrophic cardiomyopathy (HCM), which is an inherited autosomal dominant disease that is caused by mutations in sarcomeric genes.2)3) Since it has been reported that enzyme replacement therapy (ERT) is effective in reducing and clearing glycosphingolipid accumulation with subsequent improvement in cardiac function, early recognition of Fabry disease has become increasingly relevant.4)5)
Here we report a case of Fabry disease which was first suspected based upon echocardiographic findings of LVH.
Case
A 44-year-old man with no history of hypertension or diabetes mellitus was admitted with sudden onset palpitations of 2 hours duration. He had been diagnosed with end-stage renal disease (ESRD) of unknown etiology at age 33 followed by renal transplantation at age 35. Since that time, he had been treated with oral immunosuppressive agents, including prednisolone (5 mg daily), mycophenolate mofetil (1,000 mg, bid) and cyclosporine (150 mg, bid).
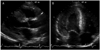
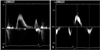
Vital signs upon arrival included a blood pressure of 150/90 mmHg and a regular rapid pulse rate of 150 bpm. The initial electrocardiogram (ECG) revealed a narrow QRS tachycardia of approximately 150 bpm with absent P waves (Fig. 1A). The patient was diagnosed with reentrant paroxysmal supraventricular tachycardia (PSVT) and was treated with intravenous adenosine. Following normalization of rhythm, the ECG showed LVH with a strain pattern, ST-T changes in leads II, V3, and V4, and a short PR interval (Fig. 1B). Chest radiography demonstrated mild cardiomegaly (Fig. 2). Laboratory studies revealed that hemoglobin was 13.3 g/dL, creatinine 1.7 mg/dL, BUN 27 mg/dL, sodium 134 mEq/L, and potassium 4.1 mEq/L. On hospital day two, transthoracic echocardiography revealed severe concentric LVH (septum 28 mm, posterior wall 20 mm) without left ventricular outflow tract obstruction, mimicking non-obstructive HCM (Fig. 3). Systolic left ventricular function was preserved but diastolic dysfunction was present. Pulsed-wave Doppler recording of mitral inflow revealed a phase resembling a normal diastolic filling pattern, with an E/A ratio of 1.6 (Fig. 4A). The mitral annulus velocity was obtained from the septal side of the mitral annulus using Doppler tissue imaging to distinguish a true normal from a pseudonormal pattern (Fig. 4B). The early diastolic tissue Doppler velocity (Ea) and the E/Ea index were 0.5 cm/s and 22, respectively, indicating increased LV filling pressure and a pseudonormal pattern. Since infiltrative diseases could not be ruled out based on clinical and echocardiographic findings, a careful history was obtained, revealing chronic hypohidrosis and heat intolerance. Fabry cardiomyopathy was thus suspected due to its typical history of early onset ESRD and renal transplantation prior to 40 years of age as well as hypohidrosis and heat intolerance. This diagnosis was confirmed by demonstration of a low plasma α-Gal A activity of 0.17 nmoles/hr/mL (normal mean, 8.5±2.4 nmol/hr/mL). A His46Arg missense mutation in the α-Gal A gene, which resulted in the substitution of arginine for histidine, was subsequently identified from lymphocyte DNA (Fig. 5). A transplant renal biopsy was performed because chronic renal allograft rejection was suspected given the increased serum creatinine level and high resistive index of the transplanted kidney; however, the biopsy was unremarkable. Enzyme replacement therapy with recombinant α-Gal A was started on an outpatient basis.
Discussion
Hundreds of inborn errors of metabolism have been reported and have been classified according to the type of metabolism involved. Lysosomal storage disorders, one of the major categories of inborn errors of metabolism caused by a deficiency of specific lysosomal enzymes, are generally classified according to the involved substrates as follows: sphingolipidoses, glycoproteinoses, mucolipidoses, mucopolysaccharidoses (MPSs), and many others. Cardiac disease is particularly prominent in lysosomal glycogen storage diseases (Pompe and Danon disease), MPSs and in glycosphingolipidoses (Fabry disease). Among lysosomal storage disorders, enzyme replacement therapy for the treatment of Gaucher disease, Pompe disease, Fabry disease and MPS I, II and VI has been approved in the United States.6)
Fabry disease is an X-linked recessive inborn error of glycosphingolipid catabolism due to deficient or absent activity of the lysosomal enzyme, a-Gal A. Cardiac involvement, consisting of progressive deposition of globotriaosylceramide (Gb3) in myocardial cells leading to LVH, is the most frequent cause of death.1) Among patients with late-onset HCM, Fabry cardiomyopathy was diagnosed in up to 6% of men and up to 12% of women.7)8) Cardiac accumulation of Gb3 leads to thickening of the cardiac wall and may cause mitral valve prolapse, arrhythmia, heart attack, heart failure and stroke. Rarely, the heart may be the only organ involved in the so-called "cardiac variant," wherein patients present with a mild, late-onset disorder limited to the heart.9)10)
Here we report a case of Fabry cardiomyopathy with early onset ESRD and renal transplantation. To our knowledge, this is the first report of Fabry cardiomyopathy in Korea. The patient's clinical and echocardiographic findings were suggestive of an infiltrative disease as well as HCM; however, HCM was thought to be more likely than infiltrative cardiomyopathy in which electrocardiography commonly exhibits a low voltage.11) In addition, the patient had typical manifestations of Fabry disease, including hypohidrosis and heat intolerance, which were eventually diagnosed both biochemically and genetically. Mild left ventricular diastolic dysfunction is commonly present in the early stages of the disease; however, in advanced stages, systolic and severe diastolic dysfunction are present.12-14) In our patient, transthoracic echocardiography revealed concentric LVH and stage 2 diastolic dysfunction. Upon initial presentation, the chief complaint was palpitations with an ECG indicative of PSVT. The prevalence of reported persistent atrial fibrillation, paroxysmal atrial fibrillation, and nonsustained ventricular tachycardia in Fabry disease are 3.9%, 13.3% and 8.3%, repectively.15) Following chemical conversion, the ECG revealed LVH with a strain pattern, ST-T changes in leads II, V3, and V4 and a short PR interval, which are all electrocardiographic findings common in most adult patients with Fabry disease. The short PR interval may have been the result of accelerated AV conduction rather than the presence of an accessory pathway.16)
Percutaneous biopsy of the transplanted kidney was performed to rule out chronic rejection. The findings on renal biopsy were normal, indicating that there was no evidence of recurrent Fabry disease in the graft. The risk of recurrence following renal transplantation has been reported to be less than 5%.17) Both short- and long-term outcomes with kidney transplantation in ESRD patients with Fabry disease are reported to be good and comparable to those of kidney transplant recipients who developed ESRD from other causes.18)
Following discharge, our patient began enzyme replacement therapy with Fabrazyme (Agalsidase-beta). Two recombinant enzyme preparations of Agalsidase-alpha and Agalsidase-beta are now available for the treatment of Fabry disease. Several studies have reported the clearance of microvascular endothelial deposits of globotriaosylceramide from the kidneys, heart and skin with subsequent improvement in neurological, renal and cardiac manifestations of the disease as well as in quality of life.3)4)19)20) Therefore, early diagnosis is imperative for the modification of these manifestations and the disease course with enzyme replacement therapy.



 XML Download
XML Download