

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
There are many established and proposed bio-molecular markers for cardiovascular disease. These bio-molecular markers include vasoactive substances such as catecholamines, endothelins, natriuretic peptides, adrenomedullin, prostaglandins, nitric oxide, and components of the renin-angiotensin system; substances related to inflammation and oxidative stress, such as C-reactive protein (CRP), interleukin-6, homocysteine, and reactive oxygen species; substances related to glucose and lipid metabolism, such as insulin, leptin, and adiponectin; and substances involved in tissue structure and remodeling, such as matrix metalloproteinases, transforming growth factor-β, and tumor necrosis factor-α.
The mammalian natriuretic peptide system is comprised of three homologous peptides-atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and C-type natriuretic peptide (CNP) and two biologically active receptors that have guanylyl cyclase (GC) activity.1) ANP and BNP bind natriuretic receptor-A (GC-A), while CNP specifically acts on natriuretic receptor-B (GC-B). Both ANP and BNP are most abundantly expressed in the heart: ANP is mainly secreted from the cardiac atrium, while BNP is mainly secreted from the cardiac ventricle. CNP, which was originally isolated from brain extracts, is synthesized in vascular endothelial cells. It was recently revealed that CNP is also produced in and released from cardiac ventricular cells.
Adrenomedullin (AM) is a potent vasodilator peptide that was originally discovered in human pheochromocytomas.2) It is widely distributed in various organs and tissues, including the cardiovascular system.
In this review, we examine the biological actions of ANP, BNP, CNP, and AM on the cardiovascular system and focus on the usefulness of these peptides as bio-molecular markers for cardiovascular disease.
Biological Actions of Natriuretic Peptides on the Cardiovascular System
The wide distribution of the GC-A receptor in various organs and tissues suggests that ANP and BNP exercise many physiological and pathophysiological functions. First, ANP and BNP have potent natriuretic and diuretic actions in the kidney. Intrarenal arterial infusion of ANP (1.0 µg/min) has been shown to increase renal blood flow, glomerular filtration rate, and urine flow in anesthetized dogs, with no change in systemic blood pressure.3) However, lower dose ANP (0.2 µg/min) produces only diuresis and natriuresis, while renal hemodynamics remain unchanged. Thus, ANP inhibits the tubular reabsorption of electrolytes, even at low doses, and induces renal vasodilation at higher doses.
Second, ANP and BNP decrease systemic blood pressure by exerting a direct relaxing effect on the vasculature. The blood pressure-lowering effect of ANP includes a shift of fluid to the extravascular compartment. Because this shift is observed in both normal and nephrectomized rats,4) it is unlikely that the fluid shift is simply attributable to ANP-induced diuresis. In addition, ANP inhibits renin, aldosterone, and vasopressin release and suppresses sympathetic tone. All these actions lead to lowering of blood pressure.
Thus, ANP and BNP released from the heart as circulating hormones have important roles in blood pressure regulation and fluid control. However, the finding that GC-A and GC-B receptors are expressed in cardiac atrial and ventricular tissues suggests that ANP and BNP, apart from their systemic actions, have certain local actions on the heart itself. In order to assess the hypothesis that endogenous ANP and BNP directly affect cardiac cell growth, we investigated the effect of a specific natriuretic peptide receptor antagonist, HS-142-1, on cardiac myocyte hypertrophy. HS-142-1 increases protein synthesis, cell size, and hypertrophy-related gene expression in cultured cardiac myocytes, indicating that endogenous ANP and BNP inhibit cardiomyocyte hypertrophy.5) In addition, endogenous and exogenous ANP suppress the proliferation of cultured cardiac fibroblasts and their ability to synthesize collagen.6) These findings suggest that ANP and BNP secreted from cardiac cells as autocrine and/or paracrine factors contribute to the inhibition of excessive cardiac cell hypertrophy and fibrosis. Indeed, mice lacking GC-A develop ventricular hypertrophy with interstitial fibrosis independent of their blood pressure in vivo.7) A human study also demonstrated that ANP administration has a suppressive effect against left ventricular remodeling after acute myocardial infarction.8)
CNP is a vasorelaxant with particularly powerful venodilator effects, contrasting with the arterial bias of ANP and BNP.9) The differential effects of ANP/BNP and CNP on arteries and veins reflect the differential expressions of GC-A and GC-B in those tissues. Although the natriuretic effect of CNP is still controversial, it does not appear to have any marked influence on renal sodium handling. Thus, the systemic hemodynamic effects of CNP are evidently less pronounced than those of ANP and BNP.
CNP is thought to have an important function as a local hormone in the vascular walls.10) As CNP is produced in vascular endothelial cells, endothelium-derived CNP can elicit direct vasodilation targeting of GC-B abundantly expressed in vascular smooth muscle cells. In addition, CNP is known to suppress growth factorstimulated migration and proliferation of vascular smooth muscle cells more potently than do ANP or BNP. CNP may play a protective role against abnormal vascular smooth muscle growth in disorders such as atherosclerosis and post-angioplasty restenosis. In fact, CNP can limit neointimal formation in arteries subjected to balloon injury, partly due to rapid re-endothelialization of the injured vessel.11)
Some in vitro studies have shown the direct effects of CNP on cardiac myocytes and fibroblasts. CNP inhibits fibroblast proliferation and extracellular matrix production more potently than does ANP or BNP.12) Endothelin-1-induced cardiac fibroblast secretion and cardiomyocyte hypertrophy are also inhibited by CNP.13) Taken together with the CNP production in cardiac fibroblasts, CNP may act as a local autocrine/paracrine regulator in both the vascular wall and in the heart.
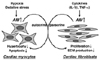
Besides their pivotal role in vascular tonus regulation and fluid homeostasis as systemic hormones, ANP, BNP, and CNP act locally to contribute to the inhibitory regulation of vascular and cardiac cell growth, including that leading to ventricular remodeling (Fig. 1).
Natriuretic Peptides as Biomarkers for Cardiovascular Disease
It is well known that plasma levels of ANP and BNP are increased in various pathological conditions, such as heart failure, myocardial infarction, hypertension, left ventricular hypertrophy, pulmonary hypertension, and renal failure.1)
It has been demonstrated that plasma levels of ANP are elevated in patients with symptomatic congestive heart failure and in asymptomatic patients with left ventricular dysfunction. As a result, plasma BNP concentrations have been used to evaluate the severity of heart failure because plasma BNP concentrations in patients with heart failure are better correlated with New York Heart Association (NYHA) functional class, hemodynamics, left ventricular filling pressure, left ventricular ejection fraction, and pulmonary artery wedge pressure.14) Like ANP levels, plasma BNP levels are useful for identifying patients with asymptomatic left ventricular dysfunction. A number of studies have demonstrated that plasma BNP is a useful biomarker for the detection of left ventricular systolic and diastolic dysfunction both in the general population and in patients with cardiovascular disease.15)16)
Plasma natriuretic peptide levels are elevated in patients with chronic renal failure, including those on hemodialysis. The fact that BNP levels increase in correspondence to the severity of renal dysfunction suggests that the decrease in renal clearance itself may affect BNP levels in patients with renal failure. For that reason, the value of plasma BNP for diagnosing cardiac dysfunction and heart failure is considered limited in patients with chronic renal failure. However, a previous study suggested that left ventricular structural and functional alterations, rather than reduced plasma clearance, constitute the prime cause of elevated BNP levels in dialysis patients.17) We also demonstrated that plasma BNP is a reliable marker of left ventricular overload, even in patients with non-dialysis chronic renal failure (Fig. 2A).18) Furthermore, high levels of plasma BNP (≥150 pg/mL) had a powerful predictive potential for the occurrence of congestive heart failure in such patients (Fig. 2B). Plasma ANP and BNP levels are increased in right ventricular dysfunction. In patients with right ventricular pressure overload due to primary pulmonary hypertension and thromboembolism, plasma ANP and BNP levels are higher than those in patients with right ventricular volume overload due to atrial septal defect.19) Plasma ANP and BNP levels are correlated with mean pulmonary artery pressure, right atrial pressure, right ventricular end-diastolic pressure, and total pulmonary resistance in these patients.
In acute myocardial infarction, the plasma ANP level was shown to be increased at the time of admission and decreased thereafter, again exhibiting a small peak on day 2 to 3. In contrast, plasma BNP levels were significantly increased on admission and reached a peak level 12 to 24 hours after admission.20) Thereafter, the BNP level decreased and achieved a second peak on day 5 to 7, possibly reflecting left ventricular remodeling. The BNP level then decreased gradually, but still remained higher than that of controls in the fourth week. The height of the second BNP peak may be a good index of left ventricular remodeling in patients with acute myocardial infarction.
A number of studies have shown that plasma ANP and BNP are increased in patients with hypertension, particularly those with left ventricular hypertrophy. In addition, plasma BNP levels are significantly increased in subjects with left ventricular concentric hypertrophy compared with those with eccentric hypertrophy or concentric remodeling, or compared with essential hypertensives with normal left ventricular geometry.21)
Elevated levels of ANP and BNP are also prognostic indicators in patients with heart failure, acute myocardial infarction, and pulmonary hypertension, and in the general population. Medical treatment can reduce the ANP and BNP levels in parallel with hemodynamic and clinical improvement in patients with these diseases. It has been shown that BNP-guided treatment of heart failure reduces total cardiovascular events compared with intensive clinically guided (non-BNP-guided) treatment.22)
Thus, plasma levels of ANP and BNP (especially BNP) are useful and reliable biochemical markers for screening, detecting progression, optimizing treatment, and predicting prognosis in the setting of cardiovascular disease.
Elevated circulating levels of CNP are found in hypoxemia, sepsis, and chronic renal failure.9) Endothelial damage caused by hypoxia and endotoxin may provoke CNP secretion from endothelial cells. Marked elevations in plasma CNP observed in renal failure may be attributable to impaired clearance. However, plasma CNP concentrations are not elevated in essential hypertension. Several studies have reported no significant increase in plasma CNP levels in chronic heart failure, in contrast to the remarkable elevations of circulating ANP and BNP. However, Kalra et al.23) demonstrated significant cardiac production of CNP in chronic heart failure by detecting a step-up in plasma CNP concentration from the aorta to the coronary sinus. In addition, coronary sinus CNP levels were correlated with the mean pulmonary capillary wedge pressure. Although further investigation is required to clarify the pathophysiological role of CNP in the heart, it is possible that cardiac CNP acts locally but not systemically in some pathological states.
Biological Actions of Adrenomedullin on the Cardiovascular System
Systemic administration of AM elicits a potent hypotensive effect due to its vasodilatory action. Intravenous bolus injection of AM causes a rapid, remarkable, and long-lasting reduction in blood pressure in a dosedependent manner.2) This reduction in blood pressure is closely associated with a decrease in total peripheral resistance, and this decrease is concomitant with increases in cardiac output and stroke volume, probably secondary to reduced afterload.24) Furthermore, the vasodilatory actions of AM are observed not only in the systemic vasculature but also in regional vascular beds, including the renal, pulmonary, cerebral, and coronary circulation. It has been shown that the vasodilatory effect of AM is mediated by at least two different mechanisms: a direct action on vascular smooth muscle cells and an indirect effect through primary action on endothelial cells.25)
Administration of AM to experimental animals increases urine output and urinary sodium excretion in a dose-dependent manner26) in association with renal vasodilation and increased glomerular filtration rate. Low doses of AM increase natriuresis without affecting glomerular filtration rate.27) Therefore, the natriuretic and diuretic actions of AM are probably involved, at least in part, in its hypotensive effect.
AM has various direct actions on vascular cells besides its vasodilatory effects. AM clearly inhibits plateletderived growth factor- or angiotensin II-induced migration of vascular smooth muscle cells.28) AM also inhibits vascular smooth muscle cell proliferation stimulated by serum or platelet-derived growth factor. In addition to these anti-migration and anti-proliferation activities, AM inhibits serum deprivation-induced endothelial cell apoptosis29) and promotes endothelial regeneration and angiogenesis.30) These properties suggest the protective role of AM against ischemia and vascular injury.
As for the cardiac actions of AM, systemic administration of AM markedly increases cardiac output in both normal men and in patients with congestive heart failure, 31) and this effect appears to be mainly due to reduced peripheral resistance. There are discrepant findings as to the direct effect of AM on myocardial contractility (i.e., positive inotropic, negative inotropic, or no significant action).
AM is known to inhibit angiotensin II- and phenylephrine-induced hypertrophy of cultured ventricular myocytes.32) In addition, AM strongly inhibits deoxyribonucleic acid (DNA) and protein synthesis and extracellular matrix production in cultured cardiac fibroblasts, suggesting that AM may play a role as a regulator of cardiac remodeling.33) AM has also been shown to inhibit cardiomyocyte apoptosis induced by doxorubicin and ischemia/reperfusion injury.
AM is synthesized and secreted from cardiac myocytes and fibroblasts, and its production is up-regulated by stimuli such as hypoxia, oxidative stress, and several cytokines in those cells, respectively.34)35) On the basis of these observations and the protective effects of endogenous AM against angiotensin II-induced cardiac hypertrophy and fibrosis and oxidative stress-mediated cardiac damage, it may be concluded that AM plays an important role as an autocrine or paracrine modulator in some cardiac disorders (Fig. 3).
Adrenomedullin as a Biomarker for Cardiovascular Disease
Ishimitsu et al.36) first reported that plasma AM is increased in hypertensive patients. Increased levels of AM are correlated with hypertensive organ damage, such as left ventricular hypertrophy and renal dysfunction. In malignant hypertension, plasma AM levels are markedly increased compared with those seen in essential hypertension; this increase is reduced after adequate antihypertensive treatment.37) Taken together, these findings indicate that plasma AM levels are increased in proportion to the severity of hypertension and that increased AM levels may function as a defense mechanism.
There have been several reports showing that plasma AM levels are elevated in patients with heart failure. AM levels are increased in accordance with severity of NYHA classification; they are furthermore positively correlated with plasma ANP, BNP, and norepinephrine levels and negatively correlated with left ventricular ejection fraction.38) Plasma ANP and BNP levels rapidly decrease after treatment of heart failure, whereas plasma AM levels decrease relatively slowly. These findings suggest that plasma AM levels are increased in proportion to the severity of heart failure and that the mechanism of their increase may be related not only to cardiac overload, but also to other hemodynamic and neurohumoral changes.
As for whether AM is a biochemical marker for cardiovascular prognosis in subjects with heart failure and cardiac dysfunction, some studies have shown that the plasma AM level is an independent prognostic indicator of mild to moderate heart failure and ischemic heart failure with left ventricular dysfunction.39)40) In hemodialysis patients with cardiovascular disease, we demonstrated that plasma AM reflects cardiac dysfunction, excessive blood volume, and inflammation better than do ANP and BNP. Further, AM has predictive value for mortality and cardiovascular morbidity.41)
Although plasma AM levels are immediately increased after the onset of acute myocardial infarction, the levels are higher in patients with congestive heart failure than in those without heart failure.42) Since AM levels are positively correlated with pulmonary capillary wedge pressure, left ventricular end diastolic volume index, and peak plasma creatine kinase, the elevation of plasma AM in the early stage of myocardial infarction mainly reflect cardiac dysfunction in proportion to disease severity. As in congestive heart failure, plasma AM concentrations in the acute phase of myocardial infarction are reported to be an independent predictor of post-infarction mortality.43)
Elevated levels of plasma AM are also observed in atherosclerotic diseases, such as chronic ischemic stroke and stable coronary artery disease without myocardial infarction. We previously showed that plasma AM levels are increased in patients with peripheral arterial disease in association with decreased ankle-brachial index and Fontaine's stage progression.44) The plasma AM also correlated with CRP and interleukin-6 levels. These findings indicate that the elevation of plasma AM concentration in peripheral arterial disease is related to the severity of the disease and likely to vascular inflammation.
Vascular inflammation is currently recognized as playing an important role in the development of atherosclerosis. Indeed, many studies indicate that inflammatory markers such as CRP and interleukin-6 are predictors of cardiovascular disease. Thus, we tested the potential usefulness of plasma AM as a marker for vascular complications in patients with multiple atherosclerotic risk factors, comparing it to other inflammatory markers. Among AM, CRP, and interleukin-6, AM was the most sensitive marker for the detection of coronary artery disease and peripheral arterial disease.45)
We further investigated the power of plasma AM for predicting future cardiovascular events in high-risk patients. When the patients were divided into three groups by tertiles of basal levels of AM, Kaplan-Meier event-free rates were decreased according to increases in basal AM levels (Fig. 4). In addition, high plasma AM was an independent determinant for cardiovascular morbidity on multivariate analysis, and its predictive value was superior to that of CRP or adiponectin.46) Because AM comprehensively reflects vascular inflammation and injury, atherosclerotic change, systemic and myocardial ischemia, and cardiac dysfunction, plasma AM seems to be a sensitive predictive marker for future cardiovascular events.
In summary, plasma AM levels are increased in various cardiovascular diseases in proportion to disease severity. Particularly, plasma AM measurements are useful as predictive and prognostic markers of atherosclerotic vascular disease.



 XML Download
XML Download