

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Cilostazol, a [6-{4-(1-cyclohexy1-1H-tetrazol-5-yl)butoxy]-3,4-dihydro-2(1H)-quinolinone; OPC-13013}, is a 2-oxo-quinoline derivative phosphodiesterase IIIA (PDE3) inhibitor that has been in use in Korea since 1991 and has been approved in the United States for the treatment of intermittent claudication since 1999.1) Recently, many preclinical and clinical studies have demonstrated that the drug has many favorable effects on athroscleoritic vascular diseases, specifically antiproliferative effects on smooth muscle cells, anti-thrombotic effects on platelets, endothelial protective effects, and modification of lipid profiles.2) The use of drug-eluting stents (DES) has dramatically increased because of their effectiveness in reducing restenosis after stent implantation. However, the limitations of stent thrombosis and restenosis in high risk subgroups remain to be resolved. Therefore, cilostazol is emerging as an alternative drug in the DES era.
Mechanism of Action
The cyclic nucleotide PDE are enzymes that degrade the phosphodiester bond in the second messenger molecules cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP). Multiple subtypes of PDE have been isolated from the human body. The therapeutic use of PDE inhibitors, predicted as early as 1977 by Weiss and Hait, is dependent on PDE distribution in various cell types.3) The main effects of cilostazol are expressed through the inhibition of PDE III, which is found predominantly in platelets, cardiac cells, vascular smooth muscle cells, and fatty tissue or adipose cells. When the action of PDE III is inhibited by cilostazol, there is an increase in cAMP levels in these cells, which in turn leads to inhibition of platelet aggregation, vasodilation, and vascular smooth muscle cell activity, an increase in heart rate and contractile force, and an improvement in lipid metabolism. Cilostazol undergoes intensive and complete hepatic metabolism via the cytochrome P450 system. This has the potential to result in some drug interactions (i.e., with erythromycin and omeprazole). Cilostazol's half-life is approximately 10 hours, resulting in an approximately 2-fold accumulation of the drug during repeat administration.1)
Smooth muscle cells
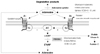
Cilostazol induces cAMP elevation in vascular smooth muscle cells via inhibition of PDE III-dependent degradation and adenosine (A2)-induced stimulation of cAMP formation (Fig. 1).4) In turn, cAMP inhibits calcium entry and migration, proliferation, and matrix synthesis in smooth muscle cells. Many downstream pathways have been suggested based on in vitro studies, including inhibition of [3H] thymidine incorporation into deoxyribonucleic acid (DNA), inhibition of interleukin-6 production, activation of cAMP-dependent protein kinase-A (PKA), induction of the p53 and p21 antioncogenes and apoptosis, downregulation of p27Kip1, and inhibition of expression of the E2F1, E2F2, and E2F4 proteins.5)6) After stent placement, cilostazol inhibits stent-induced P-selectin expression on platelets and upregulates leukocyte Mac-1, thereby inhibiting neointimal growth.7)
Endothelial cells
Cilostazol induces elevation of endothelial cAMP via inhibition of PDE III degradation and A2-induced stimulation of cAMP formation in endothelial cells. Elevated cAMP stimulates endothelial cell proliferation, reduces expression of adhesion molecules (i.e., vascular cell adhesion molecule-1), inhibits cytokine release and action (monocyte chemoattractant protein-1, platelet derived growth factor, tumor necrosis factor-α), and inhibits apoptosis.8)9) Cilostazol induces nitric oxide (NO) production through endothelial nitric activation via a cAMP/PKA- and PI3K/Akt-dependent mechanism, and this effect is involved in capillary-like tube formation in human aortic endothelial cells.10) Moreover, cilostazol has a potential anti-inflammatory effect on monocyte-endothelial interactions via upregulation of intracellular cAMP.
Platelets
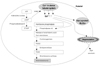
Platelets exhibit predominant PDE III reaction, with subordinate PDE II and V reaction. Cilostazol elevates cAMP in human platelets via inhibition of PDE III-dependent degradation at high concentrations. It also potentiates the cAMP-elevating and antiplatelet effects of adenylate cyclase-activating agents (prostaglandin E1 and I2) at a low concentration.5) Increased cAMP levels facilitate the influx of free calcium ions back into storage granules in the platelets. The free calcium ions are needed for the formation of the glycoprotein IIb/IIIa complex, degranulation of the storage granules containing aggretating substances, and production of thromboxane A2 (Fig. 2).
Brain cells
The anti-platelet and vasodilating effects of cilostazol are believed to underlie its preventive effect on recurrent stroke. It is also considered to be effective in promoting recovery from functional damage of the endothelium in cerebral penetrating arterioles. Cilostazol also promotes brain cell survival ascribed to maxi-K channel opening-coupled upregulation of creatinine kinase 2 phosphorylation and downregulation of PTEN phosphorylation, with a resultant increase in phosphorylation of Akt and CREB.12)
Clinical Trials and Evidence for Clinical Effects
Prevention of restenosis after vascular intervention
Cilostazol inhibits smooth muscle cell migration and proliferation and promotes endothelial cell growth. It has been viewed as a novel antiproliferative drug acting on smooth muscle cells. After its anti-restenosis mechanisms were first acertained, cilostazol was suggested as an effective agent for the prevention of restenosis after vascular interventions. Cilostazol has been proven in multiple randomized clinical trials to reduce restenosis in the setting of coronary interventions. In the balloon angioplasty era, some clinical trials suggested cilostazol as a potential agent for reducing restenosis. Balloon angioplasty plus cilostazol administration showed favorable outcomes compared to bare-metal stenting in the setting of small-vessel disease.13) After bare-metal stenting, cilostazol was comparable or superior to ticlopidine in reducing restenosis.14) On a cost-effect analysis, treatment with cilostazol proved to be a cost-saving or dominant strategy in patients with coronary bare metal stent (BMS) implantation.15)
Although the introduction of DES has dramatically reduced neointimal hyperplasia and restenosis compared with BMS, several conditions still place patients at high risk for restenosis: diabetes mellitus, long lesions, and small vessel disease.16) Combination aspirin and cilostazol therapy have been shown to reduce in-segment restenosis by 50% compared to combination aspirin and clopidogrel therapy in diabetic patients after DES implantation.17)
A similar result was demonstrated in another study documenting that triple therapy (aspirin, clopidogrel, and cilostazol) reduced in-segment restenosis by 50% compared to dual therapy (aspirin plus clopidogrel) in diabetic patients.18) In long lesions, cilostazol reduced late in-stent and in-segment loss and reduced the occurrence of target-vessel revascularization at 6 months after DES implantation.19) Furthermore, cilostazol treatment changed in-stent restenosis morphology to a more focal pattern compared to that seen in clopidogrel-treated patients.20)
Antithrombotic effects
During and after elective BMS implantation, aspirin plus cilostazol showed an antithrombotic effect comparable to that seen with aspirin plus ticlopidine.21) Cilostazol also demonstrated effectiveness comparable to clopidogrel in preventing thrombosis after bare-metal stenting.22) In the BMS era, there is no evidence to suggest that any one combination therapy-aspirin plus ticlopidine, aspirin plus clopidogrel, or aspirin plus cilostazol is superior to the others for use after cardiac events.
DESs appear to prolong the duration of re-endothelialization, and stent thrombosis has emerged as a major concern. Late stent thrombosis has been shown to be substantially higher among real-world patients compared to those included in clinical trials.23) Current American College of Cardiology/American Heart Association (ACC/AHA)/Society for Cardiac Angiography and Interventions (SCAI) guidelines recommend dual antiplatelet therapy for at least 12 months in patients at low risk for bleeding, especially off-label patients. Cilostazol is considered the third antiplatelet agent in patients at high risk for thrombosis. Diabetic patients who underwent DES implantation and who were treated with a combination of aspirin and cilostazol showed a late stent thrombosis rate comparable to that seen in patients treated with aspirin and clopidogrel during 7.1 months of follow-up.17) A randomized multicenter study comparing triple therapy (aspirin, clopidogrel, and cilostazol) and dual therapy (aspirin and clopidogrel) in patients with long lesions requiring a long DES showed similar rates of stent thrombosis at 9 months.19) Until now, there is no concrete evidence to suggest that triple therapy is superior to dual therapy in preventing stent thrombosis in the short-term. We need to gather data concerning the effect of cilostazol on very late stent throbosis (i.e, that presenting beyond 1 year after stent implantation). Another recommendation is substitutive or additive use of clopidogrel in nonresponders.
About 20% of patients undergoing percutaneous coronary intervention are resistant to clopidogrel. Despite clopidogrel therapy, these patients are at high risk for recurrent thrombotic events.24) Triple therapy attenuates the prevalance of clopidogrel resistance in patients undergoing coronary intervention.25)
Cilostazol seems to be an effective alternative or additive antiplatelet agent in patients who need to discontinue clopidogrel because of drug-related side effects, those at high risk for stent thrombosis, and those with clopidogrel resistance; however, available evidence remains limited due to small study effects, and large randomized clinical trials are needed.
Effects on peripheral vascular disease
In Korea, two agents are available for the treatment of intermittent claudication: pentoxifylline and cilostazol. Cilostazol has a vasodilating effect because it inhibits PDE III in the blood vessel by increasing cAMP levels in vascular smooth muscle cells. Cilostazol also increases nitric oxide synthesis in vascular endothelium without interfering with prostacyclin synthesis.10) The vasodilatory effect is not uniform or dose-dependent. It shows high selectivity for the vertebral and femoral arteries.26) Cilostazol therapy (200 mg/day for 6 weeks) has been shown to improve skin temperature and blood flow in the legs and feet of patients with peripheral occlusive disease.27) The clinical efficacy of cilostazol has been demonstrated in many clinical trials. It has been shown to be beneficial in improving maximal and pain-free walking distances in patients with intermittent claudication who do not have rest pain or peripheral tissue necrosis. Randomized trials have shown that the mean change in maximal walking distance is superior to that achieved with pentoxifylline; the effect is dose-responsive.28) Cilostazol also has a role in the prevention of restenosis after percutaneous transluminal angioplasty in the setting of peripheral vascular occlusion. After peripheral endovascular therapy, freedom from target-lesion revascularization and all adverse events (restenosis, amputation, and death) has been shown to be significantly higher in cilostazol-treated patients than in ticlopidine-treated ones. The American College of Chest Physicians evidence-based clinical practice guidelines (8th edition) for antithrombotic therapy for peripheral artery occlusive disease recommends cilostazol in patients with moderate-to-severe intermittent claudication who do not respond to exercise therapy and who are not candidates for surgical catheter-based intervention (Grade 1A).29)
Risk of Bleeding
In many clinical trials in patients undergoing percutaneous coronary intervention (PCI), triple therapy has not been shown to increase bleeding event rates, despite its antithrombotic effects.18)19) A secondary prevention trial of stroke showed that cilostazol reduced the risk of stroke without increasing the risk of bleeding complications in Japanese patients.30) Why does a more potent antiplatelet therapy not increase the risk of bleeding? Although the precise underlying mechanism is unknown, one hypothesis is that cilostazol does not interrupt the inital process of thrombus formation (i.e., adhesion of platelets to the vascular wall).5) The addition of cilostazol to aspirin, clopidogrel, or the combination of the two leads to no additional increase in bleeding time.
There are two important safety concerns considered by the Food and Drug Administration (FDA)'s Cardio-Renal Advisory Committee; adverse survival effects in heart failure and lack of information on the use of the cilostazol in combination with clopidogrel. Other drugs with PDE III inhibition properties-notably milrinone, vesnarinone, and enoximone-have been shown to increase mortality in severe heart failure patients {New York Heart Association (NYHA) class III and IV}. In an analysis of trials excluding patients with acute heart failure, the risk of cardiovascular events and mortality associated with cilostazol was found to be similar to that of placebo. However, there is no data concerning the effects of cilostazol on patients with heart failure. Cilostazol is contraindicated in patients with congestive heart failure of any severity.
Future Perspectives and Conclusions
Cilostazol has pleotropic effects, including inhibition of neotitimal hyperplasia and antithrombotic effects. It holds the promise of preventing restenosis and stent thrombosis. However, at the present time, we need more data in order to answer the following questions: 1) What is the optimal combination, and for how long should antiplatelet agents be used after DES implantation? 2) Who is an optimal candidate for cilostazol treatment among coronary artery disease patients? 3) Would cilostazol treatment be beneficial with regard to cardiovascular events and mortality?
In conclusion, based on laboratory and clinical studies, cilostazol is an effective adjunctive phamacologic agent for the prevention of stent thrombosis and instent restenosis in the DES era. Additional data are needed in order to determine risk-benefit relations in patients with atherosclerotic coronary artery disease.
 XML Download
XML Download