

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
The renin-angiotensin system, acting through the major effector peptide angiotensin II, has potent effects on the blood pressure, the water and sodium homeostasis and the end-organ damage in the heart, vessels, brain and kidneys.1-3) Consequently, inhibiting the RAS has been an important therapeutic strategy for the treatment of hypertension and the related end-organ damage.4-7) Although the RAS has been studied for more than a century, our understanding of its mechanisms remains a bit incomplete. ACE inhibitors and angiotensin II type 1 receptor blockers(ARBs) have many other beneficial effects in addition to decreasing the blood pressure.8-10) The transmembrane protease ACE2 has recently emerged as a negative regulator of the RAS as it counterbalances the multiple functions of ACE.11)12) This review will explore the molecules and pathways that are involved in the RAS, and we will identify potential novel therapeutic targets for cardiovascular diseases.
Renin
Professor Robert Tigerstedt, a noted Finnish physiologist, discovered the renin system in 1898 at the Karolinska Institute in Sweden.13) Renin catalyzes the first, rate-limiting step in the RAS. Renin is an aspartyl proteolytic enzyme and its substrate is angiotensinogen.14) Renin cleaves a leucine-valine bond in the N-terminal region of human angiotensinogen or it cleaves a leucine-leucine bond in the angiotensinogens of other species to produce the decapeptide angiotensin I. Angiotensinogen is a large globular protein that's mainly derived from the pericentral zone of liver lobules. Juxtaglomerular cells(JG) cells of the afferent arteriole of the kidney are the locus for renin synthesis, storage and release. Translation of renin mRNA in these cells produces pre-prorenin, which in turn is then converted to prorenin by the removal of a single peptide and glycosylation. Some of the prorenin to renin conversion takes place in the juxtaglomerular cells, and both renin and prorenin are secreted from these cells. Prorenin is the more abundant circulating form of renin.15-17) Active renin is secreted in response to four regulatory mechanisms: the renal baroreceptor, the macula densa and the beta1-receptor stimulation via renal nerves and humoral factors. An intrarenal vascular stretch receptor in the afferent arterioles stimulates renin secretion in response to reduced renal perfusion pressure. When the macula densa senses a decrease of the distal tubular salt delivery, then a feedback signal initiates a series of steps that ultimately stimulate renin secretion. The JG cells are directly innervated by sympathetic nerves. Direct stimulation of the renal nerves increases renin's release from the JG cells through a beta1-adrenergic mediated mechanism. Many humoral factors have also been implicated in the secretion of renin. The primary stimulatory intracellular "second messenger" for renin release is the cyclic nucleotide cAMP, which is probably the second messenger for stimulation via the beta-adrenergic nerves and prostaglandins.18)
Developing a renin inhibitor has been marred by such issues as potency, bioavailability, duration of action and the cost of synthesizing it. Aliskiren is the first representative member of a new class of non-peptide, orally active renin inhibitors. Clinical studies on hypertension patients have recently demonstrated dose-dependent reductions of the blood pressure with administering Aliskiren, and the drug's safety and tolerability were similar to those of the ARBs.19)
Angiotensins
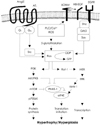
Angiotensins are peptide hormones that are produced by a series of proteolytic reactions starting with the cleavage of angiotensinogen by renin. Angiotensin I has no known biologic role aside from serving as the precursor of angiotensin II and III.20) Angiotensin II is the key effector hormone of the RAS, and it causes vasoconstriction, increased afterload and the retention of sodium and fluid. All of these actions affect the cardiovascular system and increase the blood pressure.21) Angiotensin II has been shown to stimulate VSMC growth, increase the expression of enzymes that produce mediators of inflammation such as phospholipase A2, and NAD(P)H oxidase, stimulate the JAK/STAT pathway and activate gene transcription of such proto-oncogenes as c-fos.22-25) Angiotensin II is produced both systematically and locally in the vessel wall by the actions of ACE, which cleaves angiotensin I to form angiotensin II. The importance of small GTP-binding proteins(G proteins) in angiotensin II signaling is becoming much clearer.2) Small G proteins are monomeric G proteins with a low molecular weight of between 20 to 40 kDa. A small G protein acts as a molecular switch that cycles between its inactive GDP-bound and active GTP-bound forms. Through the activation of small G proteins such as Ras, Rho, and Rac, angiotensin II induces vascular smooth muscle cell remodeling, including proliferation, migration and hypertrophy of the smooth muscle cells(Fig. 1).2)26-29) Although the circulating angiotensin II levels decrease in response to acute ACE inhibitor treatment, the circulating angiotensin II levels tend to increase in patients who are taking ACE inhibitors over long periods.30) The formation of angiotensin II, despite achieving effective ACE inhibition, may be explained by the activity of alternative enzymes that are capable of catalyzing the conversion of angiotensin I to angiotensin II. One such enzyme, which may be important in humans, is human heart chymase. Other enzymes known to catalyze this conversion include cathepsin G, and trypsin.31) In humans, direct comparison of the renal hemodynamic response to angiotensin II antagonist and ACE inhibitors has suggested that 30% to 40% of generating angiotensin II is non-ACE dependent when the renin system is stimulated by a low-salt diet. This percentage is even higher when the renin system is suppressed by a high-salt diet.32)
Data from the current well-documented studies on RAS's role in the development and maintenance of arterial hypertension has implicated angiotensin II in cardiovascular and renal pathologies, and these include cardiac left ventricular hypertrophy(LVH) and structural alterations of the vasculature, heart and kidney(e.g., neointima formation, post-infarct remodeling and nephrosclerosis).33) In this regard, angiotensin II has been recognized as a growth-promoting factor that contributes to structural alterations in various organs.34-37) Angiotensin III [Angiotensin(2-8)] is more lipid-soluble than is angiotensin II, and it is found in relatively high concentrations in the brain and cerebrospinal fluid. Angiotensin III is less potent than angiotensin II for eliciting biological responses. This is due both to the lower affinity of angiotensin III for the angiotensin II binding sites and also to the more rapid degradation of angiotensin III. Angiotensin IV [Angiotensin(3-8)] has effects on the brain that suggest it has a role in memory. It also stimulates the release of plasminogen activator inhibitor from endothelial cells.38) Angiotensin(1-7) is a weak agonist that binds with low affinity to the AT1 receptor, so the net effect of large amounts of that peptide is to inhibit the action of angiotensin II at that receptor. In addition, angiotensin(1-7) induces formation of nitric oxide and vasodilator prostaglandins, and it retards vascular smooth muscle growth via a mechanism that does not involve the AT1 receptor.39)
ACE and ACE2
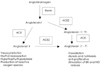
Angiotensin-converting enzyme(ACE) is a zinc metallopeptidase that's widely distributed on the surface of endothelial cells and epithelial cells.40) Because of its dual role in regulating the levels of angiotensin II and bradykinin, the positive clinical effects of ACE inhibitors were thought to be the consequence of concomitant reductions in the production of angiotensin II and the degradation of bradykinin(Fig. 2).40)41) Recent evidence has suggested that some of the beneficial effects of ACE inhibitors on cardiovascular function and homeostasis can be attributed to other mechanisms. These include the accumulation of the ACE substrate N-acetyl-seryl-aspartyl-lysyl-proline, which blocks collagen deposition in the injured heart, as well as the activation of the ACE signaling cascade, which involves the activation of kinase CK2 and the c-Jun N-terminal kinase in endothelial cells, and this leads to changes in gene expression.30) It is clear that ACE inhibitors represent one of the major advances for cardiovascular therapeutics over the past 20 years (Table 1). They have proved effective for treating hypertension, they decrease the mortality of patients with congestive heart failure and left ventricular dysfunction after myocardial infarction, and they delay the progression of diabetic nephropathy.42) The development of hypertensive left ventricular hypertrophy(LVH) is an early and important finding that may have direct pathophysiological implications on the progression from early hypertension to cardiovascular death.43) It is well established that a reduction in blood pressure will reduce hypertensive LVH.44) However, experimental evidence has suggested that various classes of antihypertensive agents have different effects on the left ventricular mass. A meta-analysis of the randomized controlled echocardiographic trials in humans, with the trials having a duration of 6 months or more, found that there were greater reductions in the left ventricular mass with administering ACE inhibitors than with administering beta-blocker, calcium antagonists or diuretics.45)
The plasma ACE levels are stable when they are measured repeatedly in the same individual, whereas large inter-individual differences of these levels have been observed. This suggests that there is strong long term control of the ACE plasma levels, and this control may possibly have a genetic origin. Studies on the structure of the human ACE gene have revealed a polymorphism involving the presence(insertion, I) or absence(deletion, D) of a 287-bp sequence of DNA in intron 16.46-49) The mean ACE activity levels in DD carriers were approximately twice that found in the individuals with the II genotype. Some investigations have suggested an association of the D allele with an increased risk for hypertension, myocardial infarction or diabetic nephropathy, and the I allele has been associated with enhanced performance in athletes.12)41)
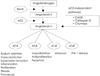
Genome-based strategies have identified a homologue of ACE with 42% sequence homology. Like ACE, this enzyme, which is named ACE2, is also a zinc-containing membrane-associated exopeptidase.12) ACE2 cleaves only a single C-terminal amino acid. It converts angiotensin II to angiotensin(1-7) and angiotensin I to angiotensin(1-9). ACE2 was initially found to be expressed in endothelial cells of the heart and in the tubular epithelial cells of the kidney. Subsequent studies have shown that the ACE2 gene expression also occurs in the gastrointestinal tract and to a lesser extent, in other organs such as lungs. Angiotensin(1-7) appears to be the main effector peptide of ACE2, and it has vasodilatory, natriuretic and antiinflammatory properties. A new regulator has entered the established metabolic RAS pathways with the discovery of ACE2(Fig. 3).19)
Angiotensin II Receptors
Angiotensin II binds to at least three known high-affinity receptor subtypes that are termed AT1, AT2 and AT4, and this converts the interaction with angiotensin II into a cellular response via signal transduction. The AT1 and AT2 receptors have similar affinities for angiotensin II, but they have distinctly different effects.1) The AT1 is the major angiotensin II receptor expressed in adults. Angiotensin II binds to the AT1 receptor and this activates G protein-coupled phospholipase C(PLC). PLC is an essential enzyme for transmembrane signal transduction by generating second messenger molecules like inositol triphosphate(IP3) and diacylglycerol(DG). IP3 induces the release of Ca2+ from internal stores, and DG is the physiological activator of protein kinase.17)48) On the other hand, angiotensin II binding to the AT2 receptor activates a counterregulatory pathway to induce vasorelaxation via activation of the kinin/NO/cGMP system.49)50) Distribution of the AT1 receptor in the adult is ubiquitous, including the vasculature, kidney, adrenal gland, heart, liver and brain. With some exceptions, most of the pathologic cardiovascular effects of angiotensin II are mediated through the AT1 receptor: hypertension, coagulation, inflammation and vascular smooth muscle cell growth.2) Several AT1 receptor blockers have become available for the treatment of hypertension or congestive heart failure(Table 2). In contrast, the AT2 receptor is mostly involved in developmental processes in the fetus; it is abundant in the fetal brain, kidney and other sites. The AT2 receptor reappears in adults, and it generally acts as a counterweight to the pathologic effects of AT1; it inhibits angiotensin II-induced growth and proinflammatory activity, and it decreases the blood pressure. The AT1 and AT2 receptors have both been cloned. They belong to the superfamily of G-protein-coupled receptors that contain 7 transmembrane regions. Their amino acid sequence seems to be highly conserved across species and across the tissues within a species.50) AT1 and AT2 receptors share only 34% homology and they have distinct signal transduction pathways. In rodents, AT1 receptor have been further subdivided into AT1A and AT1B.51) In amphibians and in neuroblastoma cell lines, an angiotensin II receptor that was not inhibited by either losartan or PD 123177 has been classified as AT3. Evidence suggests that the AT4 receptor, via angiotensin IV [angiotensin(3-8)], is an important mediator of the expression of plasminogen activator inhibitor-1(Fig. 4).51)
Conclusion
This review summarizes the molecules and pathways of the RAS. Therapeutic agents that block the RAS at several points along its pathway have significantly reduced the cardiovascular and renal morbidity and mortality. Both ACE and ACE2 are involved in the production of the biologically active peptides angiotensin II and angiotensin(1-7) from angiotensin I. There are currently ongoing studies that will elucidate the physiological roles of ACE2 in myocardial function and its contribution to kidney damage. It still remains to be seen whether more complete blockade of the RAS will improve the clinical outcomes of patients suffering with cardiovascular diseases.



 XML Download
XML Download