

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Peripheral nerves undergo Wallerian degeneration following division, crush injury, interference of the blood supply or other types of injury to a nerve, and this may be followed by neurilemma cell proliferation and axonal regeneration.1) The regeneration effort is triggered by the reexpression of nerve growth factor (NGF) or other neurotrophic factor genes in the non-neuronal cells around the site of injury.2) The vertebrate hearts are normally well innervated by autonomic nerves.3) Myocardial infarction(MI) causes cardiac nerve injury and denervation,4) and this may be followed by nerve sprouting in both animals and humans.5-8) Zou et al.9) recently reported that there is an upregulation of NGF protein and its mRNA expression 1 month after MI in a canine model. However, NGF may not the only neurotrophic factor in the heart. Other neurotrophic factors may also play a role in cardiac nerve sprouting. Furthermore, it is unknown whether or not there is persistent nerve sprouting beyond the first few months after MI. To determine which neurotrophic genes are involved, how long the nerve sprouting persist and whether or not the nerve sprouting activity is heterogeneous, we performed DNA microarray studies on mice hearts that were harvested from 3 hours to 2 months after MI. The results were then confirmed by performing quantitative mRNA analyses. We also kept 6 mice alive from 3 to 13 months after MI to determine if there was persistent nerve sprouting. The architectural arrangement of ventricular muscle mass resembles that of a Gordian knot,10) with an outer(horizontal) loop and inner(vertical) loop forming a helical structure. Both the sequence and the force of the contraction in this helical structure may be determined by the cardiac innervation. While this new concept of ventricular architectural arrangement is considered important when determining ventricular function,11) there is no data on the distribution of cardiac nerves in the helical structure. Because of its small size, we were able to mount the entire cross section of a mouse heart on a single slide to determine if there is differential nerve density in the outer and inner loops. The purpose of the present study is to use the mouse MI model to test the following hypotheses: (1) MI induces persistent neurotrophic factors overexpression and nerve sprouting. (2) The nerve sprouting activity is temporally and spatially heterogeneous.
Materials and Methods
The study protocol was approved by the Institutional Animal Care and Use Committee, and it followed the guidelines of the American Heart Association.
First(survival) surgery for creation of mi in mice
The FVB mice were obtained from the Jackson Laboratory. The mice were anesthetized intraperitoneally with an injection of ketamine and xylazine. The hair on the chest and that overlying the trachea was clipped and the skin was prepared with betadine and alcohol. The neck was opened with using sterile techniques. Direct trachea visualization was needed to guide an endotracheal tube(polyethylene tube, size 60) from the mouth into the trachea. A volume-cycled rodent respirator(Harvard Co) was used to provide positive pressure ventilation at 200-300 µL/cycle and a respiratory rate of 120 cycles/min. After the thoracic cavity was opened, ligation of the left coronary artery was performed with a 7-0 silk suture that was placed 3 to 4 mm from the tip of the left auricle. A group of mice was sacrificed 3 hours after MI. In the remaining mice, the chest was closed with continuous 6-0 prolene suture. We then used PE 50 tubing and a syringe to evacuate the air from the pleural space, and this was followed by 4-0 polyester suture to close all the skin wounds. The animals were then extubated. The skin overlying the trachea was also closed with placing one 4-0 polyester suture. The mice were kept warm by a heat lamp or heating pad and they were watched until they were fully recovered from anesthesia and surgery. Buprenorphine 0.05-0.1 mg/kg was given subcutaneously for postoperative analgesia. Among the 40 mice with MI, 8 died of surgical complications. The remaining 32 MI mice were used for data analyses.
Second surgery for organ harvest, inspection and histological analysis
Because the mice hearts were small, it was difficult to perform both immunohistochemical studies and microarray analyses using the tissues from the same heart. Therefore, 15 MI hearts were used exclusively for microarray analyses and 17 MI hearts were used exclusively for immunohistochemical studies. The chests of the mice were opened under general anesthesia and the hearts were quickly excised. The hearts that were used for microarray analyses were immediately frozen in liquid nitrogen and stored in -80℃ for later processing (see below). The hearts that were used for immunostaining were fixed in 4% formalin for 45 min to 1 hour, followed by storage in 70% alcohol.12)
mRNA Analyses
The total RNA was extracted from frozen tissues using TRIZOL(Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. The RNA was treated with Dnase I(Qiagen) to degrade any trace of DNA, and later it was cleaned again with using a Rneasy Kit(Qiagen). The concentration and integrity of RNA were evaluated by an RNA 600 Assay kit(Agilant Technology). The total RNA was reverse transcribed and amplified by TaqMan Reverse Transcription Reagents(Applied Biosystems, Foster City, CA, USA). The expression levels of the candidate genes were measured by real time qRT-PCR(quantitative RT-PCR) on an ABI PRISM 7700 Sequence Detection System(Applied Biosystems) with using SYBR Green PCR Master mix(Applied Biosystems) and primer pairs that were specific for canine. For each assay, we used GAPDH and the target gene from the same samples, along with the relevant standard samples, to control for amplification. The mRNA levels of the target genes were calculated using the relavent standard curves, and these levels were then divided by the mRNA level of the GAPDH obtained from the same samples to derive a normalized value for each sample. The amplified cDNA fragments were size-fractionated on 1.7% agarose gels and visualized by ethidium bromide staining. The sequences of the studied genes were obtained from GenBank and the primers were designed with the aid of Primer Express™ software(Applied Biosystems).
DNA microarray analyses
An affymetrix array(Mouse Expression Array 430A) was used for the mRNA expression profiling(transcriptomics). The experimental procedures for the gene chips were performed according to the Affymetrix Gene Chip Expression Analysis Technical Manual. RNA was isolated from tissue using a Qiagen RNeasy Mini kit(Qiagen, Valencia, CA). Double-stranded cDNA was synthesized using the MessageAmp aRNA kit and an oligo-(dT)24 anchored T7 primer(Ambion, Austin, TX). Two samples(duplicate) of the 2.5 µg of total RNA from each sample were used to start the cDNA synthesis. Biotinylated RNA was synthesized using the Message-Amp aRNA kit and an oligo-(dT)24 anchored T7 primer(Ambion, Austin, TX). The biotinylated RNA products were purified using Qiagen RNeasy columns and then fragmented to a size of 30 to 200 nt. Ten µg of the biotinylated fragmented RNA was then hybridized with Affymetrix GeneChip arrays(Mouse Expression Array 430A). After washing, the arrays were stained with streptavidin-phycoerythrin(Molecular Probes, Eugen, OR); the signal was amplified by biotinylated anti-streptavidin(Vector Laboratories, Inc., Burlingame, CA) and then scanned on an Agilent Genearray scanner. The intensity for each feature of the array was captured with Affymetrix GeneChip Software(MAS 5.0), according to the standard Affymetrix procedures. The abundance of the mRNA was determined based on the average of the differences between the perfect match and intentional mismatch intensities for each probe family. Gene induction or repression was determined using the appropriate statistical packages(such as Silicon Genetics' GeneSpring 5.0, Affymetrix DMT 3.0).
Immunohistochemical studies
Samples were taken from the infarcted site and non-infarcted LVFW and they were routinely processed. Five micron thick sections were mounted on charged slides. A modified immunohistochemical ABC method was used for immunostaining for GAP-43(a marker of nerve sprouting), TH and neurofilament(a markers of sympathetic nerves).12) The density of the stained nerves was determined using ImagePro software and this was expressed as the nerve area divided by the total area that was examined(µm2/mm2).13) The investigators were kept "blind" to the specimen's source. The nerve density of each slide was determined by the average of 3 fields that had the highest nerve density.
Statistical analysis
The data are presented as means±SDs. We performed ANOVA to determine the importance of the length of time from the MI(11 time points, including the control) and the location(peri-infarct versus remote sites) on the nerve density(the dependent variable). We also separately counted the nerve densities in the inner loop and outer loop at 6 time points, including the control. These sections were taken from the non-infarcted myocardium. Multivariate ANOVA was also used to determine the importance of time and location on the nerve density. A p value of ≤0.05 was considered significant.
Results
All the hearts included in the MI groups had ST segment elevation on the surface ECG during the first surgery(Fig. 1A); there was the presence of visible aneurysm during second surgery and evidence of MI according to the histological examinations(Fig. 1B). After the heart was removed, we separated the tissues from the peri-infarct region and the remote region according to the example shown in Fig. 1B.
Time course and spatial distribution of nerve sprouting activity after MI
Acute MI resulted in cardiac nerve sprouting within 3 hours. Active nerve sprouting was most apparent within 1 week after MI. The nerve sprouting activity returned to a normal level 1 month after MI in the peri-infarct region and 3 months after MI in the remote region.Fig. 2 shows the histological sections of a heart harvested within 3 hours of MI. Panel A shows the suture tracks(green arrows) and acute necrosis. The peri-infarct sites(B and C) have more GAP-43 immunopositive nerve structures than did a remote site(D) at this time point. The densities of GAP-43 immunopositive nerve fibers peaked 3 hours after MI, and this increased by >5 times that of the control level at the peri-infarct area and >3 times that of the control level at the remote area. The nerve fiber density then progressively declined over the 2-month study period in all the mice we studied(Fig. 3). Multivariate regression analyses showed that the time from MI was a significant factor that determined the nerve density(p<0.0001). There was no sites and the remote sites.
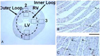
Differences between the outer and inner loops
We observed that GAP-43 immuno-positive nerve fibers were more concentrated in the outer loop than in the the inner loop in the normal control mice(Fig. 4). Similarly, MI-induced nerve sprouting was also more apparent in the outer loop than in the inner loop. Fig. 5 shows the nerve densities of all the mice we examined. Multivariate ANOVA analyses showed that both time from MI and the location(outer versus inner loop) were significantly related to the nerve density(p=0.0002 and p<0.0001, respectively).
mRNA of the Nerve Specific Markers
GAP-43 is expressed in sprouting axons, and it may not be expressed in the mature nerve. To determine the total nerve density and the density of the sympathetic nerves, we attempted to perform immunostaining for synaptophysin(SYN) and tyrosine hydroxylase(TH); however, the quality of the immunostaining was not optimal for quantitative analysis. So, we used qRT PCR to measure the mRNA of TH and SYN in the peri-infarct and remote areas. Fig. 6 shows the results. Both the TH and SYN mRNA were significantly(>1.5X) increased in the peri-infarct areas over the entire study period, except for at 1 month after MI(Panel A). In the remote areas, the TH mRNA was increased temporarily(at 3 hours and 3 days) and then it was increased again at 1 month after MI. There was no change of SYN in the remote sites. This data suggests that spouting nerve fibers(GAP-43 expressing nerve fibers) were more likely to form adrenergic synapses in the peri-infarct areas than in the remote areas. These data also suggests there was increased sympathetic innervation up to 2 months after MI in the peri-infarct areas.
Expression of neurotrophic factors in the myocardium after MI
We used microarray to screen the mRNA expressions of neurotrophin, neuropoietin, various growth factors and interleukin, which may play roles for nerve sprouting in the myocardium after MI. These genes were selected based on a literature survey.9) Table 1 shows the gene expression as a ratio to the control. A significantly increased gene expression was defined as > 3X of the baseline level of the gene expression for at least two consecutive time points. According to this definition, NGF-β, NGF-γ, insulin-like growth factor 1-2(IGF 1-2), leukemia inhibitory factor(LIF), transforming growth factor-β3(TGF-β3) and interleukin -1α(IL-1α) were increased in the peri-infarct areas. Only NGF-γ, IGF 1-2 and TGF-β3 were increased in the remote areas. The increased NGF and LIF levels were confirmed via qRT PCR analyses(Table 1)(Fig. 6), and this was defined as >1.5X of the mRNA expressions as compared with the control.9)
Discussion
We demonstrated in the mouse model that MI induced active nerve sprouting in both the peri-infarct areas and remote areas, and this started within 3 hours and persisted for at least 1 week after MI. These changes were associated with upregulation of several neurotrophic factors(NGF, IGF 1-2, LIF, TGF-β3 and IL-1α) primarily in the peri-infarct areas; this also started within 3 hours and persisted for at least 2 months after MI. There was increased SYN and TH mRNA expressions after MI in the peri-infarct areas, but not in the remote areas for at least 2 months, suggesting that the spouting nerve fibers were more likely to form adrenergic synapses in the peri-infarct areas than in the remote areas. The outer loop of the heart had more GAP-43 immunopositive nerves than did the inner loop, and this difference was most apparent at 3 hours to 3 days after MI.
Upregulation of NGF after MI
We demonstrated active NGF upregulation occurs in this mouse model soon after MI. The NGF upregulation was shown both on DNA microarray and on the qRT PCR analyses. Compatible with these results, NGF is also elevated after MI in dogs.14) In one study, Zhou et al.9) demonstrated that the NGF mRNA was significantly upregulated within 3 days after MI and this persisted for 1 month both in the infarcted and non-infarcted left ventricular myocardium. The data from the present study showed that NGF mRNA was significantly elevated even at the earlier time point(3 hr), but this returned to normal at 1 month. The NGF level was again elevated at 2 months after MI. The different time course could be due to the differences in the species and sampling sites. However, both studies support the hypothesis that NGF is upregulated after MI.
Upregulation of other neurotrophic factors after MI
In addition to NGF, DNA microarray analyses showed that other neurotrophic factors were also upregulated after MI. We showed that the LIF was upregulated via the DNA microarray analyses. The same results were confirmed by qRT PCR. Consistent with these results, others15) have reported that LIF protein was increased by 40% at 48 hours after MI in mice. We also demonstrated by DNA microarray analyses that IL-1(was increased after MI, primarily at the peri-infarct sites. This finding was consistent with that reported by Nian et al. In our study, IL-1,16) IL-1β and IL-6 were increased at only 1 time point after MI. Gwechenberger et al.17) demonstrated with performing in situ hybridization that IL-6 mRNA was induced in myocytes in the viable border zone after 1 hour of ischemia and after 3 hours of reperfusion in canine ventricles. The present study showed that IGF-2 was consistently upregulated at all time points after MI in the peri-infarct regions, and at 3 days and 1 wk in the remote regions. These results were consistent with previous studies that showed upregulated IGF after acute MI in rat hearts.18) However, the systemic(serum) IGF-1 levels in human patients are known to be reduced in the early phase of acute MI, but a compensatory increase of IGF appears to occur by 1 year.19) The increased TGF-β we found was consistent with that reported by other stusies.20) Our results showed that brain-derived neurotrophic factor(BDNF) was increased only up to 1.93X after MI. By our definition, an increase of <3X was considered negative. However, Drapeau et al.21) recently demonstrated that BDNF can be detected in rat ventricles after MI. The <3X increase of BDNF in our study does not rule out the possibility that BDNF played a role in nerve sprouting after MI.
Beneficial effects of neurotrophic factor upregulation after acute MI
The simultaneous upregulation of multiple neurotrophic factors after MI suggests that neurotrophic factors are important in the cardiac responses to acute ischemic injury.22)23) Compatible with this hypothesis, Abe et al.14) demonstrated that intracoronary infusion of exogenous NGF protects against postischemic neural stunning of sympathetic cardiac nerves during brief myocardial ischemia and reperfusion, and that using NGF antibody to block endogeneous NGF had opposite effects. Zou et al.24) showed that injection of LIF plasmid DNA into the thigh muscle of mice immediately after inducing MI markedly attenuated the left ventricular remodeling, such as the extent of infarction and the myocardial fibrosis. Furthermore, IGF is known to have cardioprotective effects after ischemic myocardial injury in mice and swine.25)26) MI may result in myocardial denervation. 4 The myocardium may be reinnervated by upregulation of neurotrophic factors. In denervated hearts(such as those after transplantation),27) reinnervation is important for improving cardiac performance.28) These findings suggest that upregulation of neurotrophic genes might confer beneficial effects to the ischemic myocardium.
Detrimental effects of neurotrophic factor upregulation after acute MI
It is know that both exogeneous and endogeneous NGF can induce cardiac nerve sprouting and increase the incidence of ventricular arrhythmia and sudden death in canine models.8) Human transplant recipients with excessive cardiac innervation are at an increased risk of ventricular arrhythmia.8) Luisi et al.29) showed the spatial heterogeneity of the sympathetic nerve function in the hibernating myocardium of swine ventricles. This heterogeneous sympathetic innervation may contribute to the high incidence of sudden death seen in that model.30) These studies suggest that the increased expression of neurotrophic factors and increased heterogeneous innervation could also induce unwanted side effects that contribute to the increased risk of sudden death after MI.31)
Nerve distribution in a helical heart
Torrent-Guasp et al.10) demonstrated that the ventricles consist of a single myofiber band extending from the right ventricular muscle just below the pulmonary artery to the left ventricular muscle where it attaches to the aorta. In this construct, sequential activation and contraction beginning in the fibers near the pulmonary artery and spreading toward the aortic end of the band might explain the pattern of ejection and suction needed for ventricular output and filling. A recent National Heart, Lung and Blood Institute workshop suggested that this model has implications for improved understanding of the electrical, electromechanical and mechanical determinants of cardiac function.25) In the present study, we demonstrated that the outer loop of this helical structure has a much higher nerve density than the inner loop in mice hearts. This difference was exacerbated after anterior wall MI that involves the apex, but not the base. Sympathetic activation in these mice might result in an uneven distribution of the contractile force in favor of the basal(outer) loop, which first contracts and causes narrowing of the ventricular chambers. This compensatory mechanism might increase the systolic function and compensate for an infarcted anteroapical left ventricle. However, we do not have the necessary hemodynamic data to test this hypothesis.
Limitations
The mouse model of MI32) differs from the human model in many respects. Coronary artery ligation in the mouse usually results in a large transmural infarction, with a possibility of aneurysm formation. In contrast, the presence of collateral circulation development and the potentially protective effect of repetitive ischemic insults in the coronary artery disease of humans means that an MI may be small and non-transmural and this does not often result in aneurysm formation. Therefore, our results cannot be interpreted as being representative of the full spectrum of infarction as this occurs in humans. A second limitation is that due to the limited size of mouse hearts, we only confirmed the overexpression of NGF and LIF with using qRT PCR techniques. Other changes of neurotrophic factors were not tested with qRT PCR. However, because the DNA microarray results were also consistent with previous reports, we do not think this limitation invalidates the conclusions of the study.






 XML Download
XML Download