

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Eisenmenger syndrome is a condition that involves elevation of the pulmonary arterial pressure to the systemic level, and this is caused by increased pulmonary vascular resistance with reversal or bidirectional shunting through a large intracardiac or extracardiac congenital heart defect. Congenital heart disease represents one such disease that is commonly associated with PAH. Eisenmenger syndrome is on the extreme end of the spectrum of PAH in the setting of congenital heart disease.1)
In 1897 Victor Eisenmenger described the clinical and pathological features of Eisenmenger syndrome in a patient with a large ventricular septal defect. The current view of Eisenmenger syndrome was recognized by Paul Wood and others who, in the 1950s, described the underlying pathophysiology as "pulmonary hypertension with reversed central shunt" and they also described the clinical phenotype of Eisenmenger syndrome that includes very different cardiac malformations associated with severe PAH.1-3)
The concept of Eisenmenger syndrome is well understood, with its progressive pulmonary vascular disease and reversal of a shunt, and this represents a final pathway for several morphological cardiac defects. Nowadays, Eisenmenger syndrome can be prevented in most pediatric patients. There have been advances in imaging, surgery and intensive care facilities, and also advances in early definitive therapy for the majority of lesions associated with a risk of developing Eisenmenger syndrome, and so a progressive decrease in the prevalence of this disease may be expected. As the annual mortality rates of Eisenmenger patients are relatively low, identification of the risk factors for this disease is problematic.4)5)
However, when patients do present with established Eisenmenger syndrome, the treatment options are generally limited to palliative measures and only very selected subgroups of patients may be considered for heart-lung transplantation. Effective drugs for the treatment of pulmonary hypertension have recently become available. These therapies may also be applicable to patients with pulmonary hypertension associated with congenital heart disease, and so they will potentially improve their symptoms and prognosis. The preliminary results of applying these drugs to patients with Eisenmenger syndrome are promising. The recent advances in pharmacological therapies for pulmonary hypertension and the prospect of heart-lung transplantation for selected patients emphasize the importance of correctly selecting patients for such therapies, with a view to further improving both the quality and duration of survival.
Definition and Mechanism of Pulmonary Arterial Hypertension
PAH is defined as an elevated mean pulmonary arterial pressure(PAP) of >25 mmHg at rest or 30 mmHg on exercise(Table 1).6) PAH has traditionally been classified according to the presence or absence of an identifiable underlying cause into primary or secondary PAH, for example, PAH with congenital heart disease (Table 2).3) PAH with congenital heart disease occurs as a result of an unrepaired systemic to pulmonary communication from a congenital heart defect; thus, it might persist even though the postoperative period after corrective surgery for the defect. There are several types of congenital heart defects that are associated with an increased risk for the development of pulmonary vascular disease, e.g., ventricular septal defect, patent ductus arteriosus and atrioventricular canal defect. For patients with a large, nonrestrictive ventricular or arterial communication, the shunt volume and direction are mainly determined by the pressure difference between the systemic and pulmonary circulations. In contrast, patients with a large atrial communication may have a right-to-left shunt that is not necessarily due to the systemic or suprasystemic pulmonary arterial pressures, and the right-to-left shunting may rather reflect lower right ventricular compliance, which is a consequence of right ventricular hypertrophy. The natural course of patients with atrial communications is generally different. PAH displays high penetrance and the onset of Eisenmenger syndrome is relatively early, and this is the rule for patients with large shunts at the ventricular or arterial level. Yet the majority of patients with even large atrial septal defects do not develop Eisenmenger syndrome, and those who develop PAH often do so much later in life.
This disease complex was renamed "Eisenmenger syndrome" and it was defined as pulmonary hypertension at systemic levels that was caused by high pulmonary vascular resistance(>800 dynes · s/cm5) with reversed or bidirectional shunting through any large systemic to pulmonary communications. This classification implied irreversibility and inoperability for a subgroup of patients with congenital heart defects. Although there have been many advances in understanding of the pathophysiology of Eisenmenger syndrome and also advances in treating this disease, there is still no cure for this progressive condition once it is established, aside from lung or heart-lung transplantation, which is associated with significant morbidity and mortality.7-11)
Untreated or even treated Eisenmenger syndrome is characterized by a progressive increase in the pulmonary vascular resistance leading to right ventricular failure and death.
Three basic mechanisms cause pulmonary hypertension. These include 1) increased pulmonary blood flow into a normal vascular bed via increased cardiac output and left-to-right shunts, 2) increased resistance in the precapillary pulmonary vessels via constriction, obliteration and obstruction, and 3) abnormal resistance in the postcapillary vascular bed via pulmonary vein obstruction, left atrial obstruction(e.g., cor triatriatum and myxoma), mitral or aortic valve disease, and left ventricular dysfunction. Specific therapies can be directed at the patients' underlying diseases.5)12)
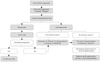
Pathophysiology(Fig. 1)
The main vascular changes of pulmonary arterial hypertension are 1) vasoconstriction, 2) smooth-muscle cell and endothelial-cell proliferation, and 3) thrombus formation in the pulmonary vessels.5-7) Thus, PAH is a dynamic and multifactorial process related to vasoconstriction and remodeling of the pulmonary vascular bed, which may be aggravated by thrombosis. These findings suggest the presence of perturbations in the normal relationships between vasodilators and vasoconstrictors, and these homeostatic imbalances are probably the consequence of pulmonary endothelial-cell dysfunction or injury.
High flow and high pressure of pulmonary artery may induce pulmonary vascular endothelial damage, leading to a loss of the endothelial barrier function. This may be associated with degradation of the extracellular matrix(activation of endogenous vascular elastase and matrix metalloproteinases) as well as to the release of growth factors(fibroblast growth factor and transforming growth factor-β). These factors induce smooth muscle cell hypertrophy and proliferation, resulting in extension of smooth muscle cells into the peripheral pulmonary vasculature, and smooth muscle cell migration with neointima formation. Furthermore, endothelial damage may result in adherence and activation of platelets and leukocytes, which favors immune inflammation and thrombosis as well as activation of the coagulation pathways.
Endothelial dysfunction also affects the production of vasoconstrictors(such as endothelin-1 and thromboxane) and vasodilators(such as nitric oxide, vasoactive intestinal peptide, and prostaglandin I2), and this shifts the balance in favor of factors that induce vasoconstriction and ultimately pulmonary vascular remodeling.
Arachidonic acid metabolites such as prostacyclin and thromboxane A2 are active in the pulmonary vessels, and these are associated with vasodilation and vasoconstriction.
Prostacyclin is a platelet inhibitor and it's capable of inhibiting endothelial cell proliferation, whereas thromboxane A2 is a platelet activator.
Endothelin-1 is a vasoconstrictor that causes smooth muscle proliferation in pulmonary vessels. Vasodilation is induced by NO, which is produced by endothelial cells through a cGMP-dependent pathway, and NO additionally inhibits platelet function and smooth vessel proliferation. In patients with PAH, there are imbalances of these potent vasoactive factors, as well other vasoactive compounds such as vascular endothelial cell growth factor, adrenomodullin, serotonin and vasoactive peptides. Patients with PAH have been found to have altered homeostatic balances of these mediators, tending towards a prothrombotic, vasoconstrictive physiology. These acquired alterations in the normal vascular physiology contribute to the onset of PAH. Other conditions like chronic hypoxemia and small vessel thrombosis can contribute to chronic changes of vasoconstriction in the pulmonary vasculature.
PAH is characterized by elevated pulmonary vascular resistance. In patients with large left-to-right shunts, the pulmonary vascular bed is subjected to ongoing shear stress. For patients with a large unrestrictive defect, the pulmonary and systemic systolic arterial pressures are the same. This leads to the histopathologic changes seen in PAH patients, which include pulmonary arteriolar medial hypertrophy, intimal fibrosis and plexiform lesions. Medial fibrosis and necrotizing arteritis changes eventually arise in the pulmonary vessels, as were described by Heath and Edwards for PAH patients with secondary congenital septal defects. It has been suggested that these histological changes may correlate with the clinical severity of PAH. For patients with Eisenmenger syndrome, these chnages may be due to the increased flow across the pulmonary vascular bed that causes peripheral extension of muscle from differentiating pericytes and intermediate cells in the precapillary vessels, as well as to damage to the pulmonary vascular endothelium, which sets in motion a series of events at the cellular level. The pulmonary vascular resistance increases with endothelial dysfunction and this ultimately causes a decrease in left-to-right shunting. Eventually, when the resistance becomes significantly elevated, the shunting becomes bidirectional and then it becomes right-to-left. The long-term follow up for those patients with elevated pulmonary vascular resistance and they have undergone closure of their defect is worse than that for the unrepaired, Eisenmenger syndrome patients with the same lesion. This may be related to the absence of a communication between the right and left circulations that may serve as a "pop-off valve" in patients who are at risk for pulmonary hypertensive crises. Normal right atrial pressures have been demonstrated in patients with Eisenmenger syndrome, suggesting that the right atrial pressure remains normal at the expense of the right to left shunt, i.e., desaturation. In addition, the systemic cardiac output is greater in patients with Eisenmenger syndrome than that in those patients with primary pulmonary hypertension, which is presumably secondary to the presence of the "pop off valve." Finally, right-sided heart failure may develop in all the patients with PAH.
For the genetic background, 6% of patients with PAH-associated congenital heart disease were found to have bone morphogenetic protein receptor type 2 missense mutations; this frequency was comparable with that reported for patients with anorexigen-associated PAH(8%), but it was considerably lower than that observed for patients with idiopathic or familial PAH(26% and 50%, respectively). This is likely to reflect the complex interrelationship between genetic susceptibility and environmental factors, such as the pulmonary blood flow or pressure, in the etiology of PAH associated with congenital heart disease.8-10)
Clinical Presentation and Evaluation
History and symptoms
For infants and children with congenital heart defect, the defects that close within the first 2 years of life are unlikely to lead to pulmonary vascular obstructive disease. Initially, infants with large unrestrictive defects usually present with signs of congestive heart failure, i.e., failure to thrive, diaphoresis and tachypnea that's caused by increased pulmonary blood flow from left to right shunting across the congenital heart defect. After years of continued exposure to high shear stress from the left to right shunting, the pulmonary vascular resistance increases to systemic levels, and this leads to reversed shunting or to right-to-left shunting; cyanosis follows and this can be severe. In rare cases, there is no history of congestive heart failure or failure to thrive in infancy, and reversal of flow may have occurred within the first 2 years of life.2)3) These patients may represent those who never had the normal physiologic decrease in the pulmonary vascular resistance or they are children with an increased susceptibility to pulmonary vascular disease, and this is especially true for children with Down's syndrome.
Chronic cyanosis results in elevated renal production of erythropoietin, and this promotes erythropoiesis and secondary erythrocytosis. Elevated hemoglobin levels represent a physiological adaptation to chronic cyanosis and they are essential to maintain adequate tissue oxygenation and prevent hypoxic end-organ damage. Increased hematocrit levels have been linked with hyperviscosity symptoms such as headaches, dizziness, visual disturbances, paresthesia and myalgia, although the data on the association between the hematocrit and viscosity is inconclusive and the data is confounded by iron deficiency.11)12) Intrapulmonary thrombosis occurs in up to a third of the adult patients with Eisenmenger syndrome, and particularly in Eisenmenger patients with large atrial septal defects, and intrapulmonary thrombosis has been associated with hemoptysis and pulmonary infarction.12-14)
In addition, most patients develop progressive shortness of breath, dyspnea on exertion and exercise intolerance. Most adult patients with Eisenmenger syndrome are highly symptomatic. More than 90% of these patients are in New York Heart Association class II or higher, and 50% of these patients report severe limitations(New York Heart Association class III or higher). These patients may also complain of chest pain.2)8)
Hemoptysis is common for patients with Eisenmenger syndrome with a reported incidence from 11% to 100%; the incidence increases with age and it is more common with advancing age. When hemoptysis occurs during the teenage years, an additional diagnostic search should be done for lung disease or left-sided heart obstructive lesions, as well as for aortopulmonary collaterals. Although it is an alarming symptom for the patient, hemoptysis seldom causes death. Furthermore, hemoptysis does not seem to be predictive of mortality.15)16)
Arrhythmias are frequent late sequelae in patients with Eisenmenger syndrome and they may pave the way for clinical deterioration, heart failure or sudden cardiac death. Eisenmenger patients have been found to have supraventricular arrhythmias on routine ECG or 24-hour Holter monitoring during long-term follow-up.3)17)
The clinical signs and symptoms of PAH are variable and they depend on underlying heart defect, the patient's age and repair status, and the degree and direction of shunting.18-20) The general symptoms that are suggestive of PAH are nonspecific and they may include breathlessness, chest pain and syncope. Central cyanosis and clubbing are the most visible clinical consequences for patients with Eisenmenger syndrome.21)22) These patients predominately exhibit cyanosis, bleeding, thrombotic diathesis, ischemic complications, an ongoing risk of bacterial endocarditis or cerebral abscess, hepatic and renal involvement, congestive heart failure and sudden cardiac death(Table 3).3)
Physical signs
On physical examination, a patient with Eisenmenger syndrome and who has not undergone surgical repair may appear cyanotic at rest or only on exertion in the early stages of disease. Particular attention should be paid for cyanosis at the oral mucosa and nail beds. In addition, clubbing of the digits may be present. Most patients have a normal jugular venous pressure on physical examination, and particularly if an atrial communication is present.
On cardiac examination, a right ventricular lift, a loud palpable single S2, a high-pitched diastolic murmur of pulmonary insufficiency and a pansystolic murmur of tricuspid insufficiency may also be present. Congestive heart failure, peripheral edema, ascites and hepatosplenomegaly may be present.
The physical findings of patients who underwent "corrective" surgery for their congenital heart defects are virtually identical to those of patients with primary pulmonary hypertension. An increase in the pulmonic component of the second heart sound and a right-sided fourth heart sound are early findings. A pansystolic murmur of tricuspid regurgitation is very common. The high-pitched diastolic murmur of pulmonary insufficiency may also be heard and this is usually relates to dilatation of the main pulmonary artery. In addition, when it is heard, a right ventricular third heart sound generally reflects advanced disease. Clubbing is not common after surgical repair. For the patient with an unrepaired atrial septal defect and Eisenmenger syndrome, there may still be fixed splitting of the second heart sound if the systolic pulmonary arterial pressure is less than the systemic systolic arterial pressure (Fig. 2).
Chest radiography
The radiographic findings are variable, and the chest radiograph may be remarkably normal for some patients. Right atrial and right ventricular enlargement with dilatation, aneurysm or calcification of the central pulmonary arteries may also be present. Enlarged central pulmonary arteries with "pruning" of the peripheral pulmonary vessels are usually late findings.
Electrocardiogram
An electrocardiogram typically demonstrates right axis deviation and right ventricular hypertrophy. A right ventricular strain pattern may be present with associated ST-T segment changes.
Echocardiography
Two-dimensional echocardiography may demonstrate any of the following: a dilated right heart, tricuspid regurgitation, pulmonary insufficiency, flattening or posterior bowing of the interventricular septum, right ventricular hypertrophy, diminished right ventricular function, and bidirectional or right-to-left shunting across the cardiac defect.23)
Cardiac catheterization
Performing cardiac catheterization in PAH patients is essential for making the diagnosis and evaluating the therapeutic options. Taking hemodynamic measurements at the time of catheterization can establish the diagnosis of PAH, and those measurement parameters for this disease are defined as a mean PAP(mPAP) greater than 25 mmHg at rest or greater than 30 mmHg with exercise, with a pulmonary capillary wedge pressure or left ventricular end diastolic pressure of 15 mmHg or less and the pulmonary vascular resistance is greater than 3 units. The cardiac output measurements are determined by performing thermodilution in patients without shunting or by the Fick method with measuring the oxygen consumption in those patients with shunting in either direction.
Pulmonary vasodilator testing
Testing at the time of catheterization with a short-acting pulmonary vasodilator is critical to deciding the therapeutic options for patients with PAH. These patients can be challenged with inhalation of 100% oxygen or by use of short-acting agents such as inhaled nitric oxide and inhaled iloprost or by intravenous(IV) adenosine, epoprostenol or prostacyclin to determine whether the vasculature is responsive to vasodilator therapy.
"Response" is defined as a reduction in the mPAP of at least 10 mmHg to achieve a mPAP of 40 mmHg or less while maintaining a normal or high cardiac output. This subset of patients may benefit from long-term medical therapy, especially with administering calcium channel blockers(CCBs). It should be noted, however, that the precise definition of a favorable acute response to vasodilator challenge is still somewhat controversial.
For testing with IV epoprostenol, an initial dose of 1-2.5 ng/kg/min is administered and this is increased by 1 to 2.5 ng/kg/min every 5 to 15 min to a maximum dose of 12 ng/kg/min. The definition of a favorable response is a >30% decrease in the PVR and/or a >10% decrease in the mPAP. An acute response to IV epoprostenol is predictive of a subsequent response to oral CCB therapy. 37% of PAH patients are considered responders.
IV adenosine produces coronary vasodilatation, decreases the systemic vascular resistance and causes relaxation of smooth muscles, including the pulmonary arteries; IV adenosine is a potent vasodilator via its actions on specific vascular receptors. Because of its short serum half-life, adenosine is a desirable agent to use as a vasodilator for the assessment of PAH. IV adenosine that's administered at a dose of 50 µg/kg/min and increased by 50 µg/kg/min every 2 min to a maximum dose of 500 µg/kg/min will acutely assess the response to a vasodilator.
Both IV epoprostenol and IV adenosine have the potential to cause a reduction in the systemic vascular resistance and they can cause systemic hypotension.
Inhaled NO testing(10 ppm) via a face mask acts as a more pulmonary selective vasodilator. A significant acute vasodilator response is defined by a fall in both the mPAP and total pulmonary resistance of >20%. Acute vasodilator testing with NO is safer than testing with CCBs.
There has been more limited data on the use of inhaled iloprost as an acute vasoreactivity testing agent. A recent study compared the response of oxygen inhalation, iloprost inhalation, IV epoprostenol and IV iloprost. IV iloprost and epoprostenol had very similar hemodynamic profiles in terms of reducing the PVR and PAP. Inhaled iloprost exerted a selective pulmonary dilatation response, resulting in a reduced PVR and PAP without systemic vasodilatation. That study suggested that inhaled iloprost should be equivalent to IV epoprostenol and inhaled NO in terms of predicting the response to CCBs.
According to previous studies, the primary objective of acute vasodilator testing in patients with PAH is to delineate the subset of patients who will respond to oral CCBs, but not to the recently developed drugs. Unstable patients or those with severe right-heart failure should not be treated with CCBs, and they need not undergo vasodilator testing.
The weight of the evidence favors either IV epoprostenol or inhaled NO as the preferred agents for vasodilator testing; IV adenosine may be used if these agents are unavailable. Testing with a short-acting agent should always take place before testing with oral CCBs, given the potential complications of vasoreactivity testing with CCBs. Only those patients who have had a substantial reduction in both the PAP and PVR with administration of an acute vasodilator should undergo further testing with CCBs.
Vasodilator testing has been best described in the setting of IPAH. Yet the literature does suggest that the pediatric population has a higher response rate to acute vasodilators. The rates of responsiveness for patients with collagen vascular diseases have been low when they were tested with inhaled NO.
Right and left heart catheterization may be required to assess the severity of pulmonary vascular disease and the "operability." The patient is considered "inoperable" if the pulmonary vascular resistance is greater than 6 units×m2 despite the administration of acute pulmonary vasodilator agents. Closing the cardiac defect of a patient with advanced disease will lead to a course similar to that of primary pulmonary hypertension patients.
Pulmonary vasoreactivity was recently shown to have prognostic value for adult patients with Eisenmenger syndrome, although it must be emphasized that the advanced therapies for PAH have a combined vasodilative and antiproliferative effect. Thus, there is also evidence to suggest that the pulmonary hemodynamics and the clinical status may improve irrespective of the response to acute vasodilator testing. Cardiac catheterization provides diagnostic and prognostic information, but the absent or a negative pulmonary vasoreactivity study should not preclude initiating disease-targeting therapy.
Angiography is performed after hemodynamic assessment, and great care should be taken to avoid precipitating a pulmonary hypertensive crisis. Selective contrast injections, preferably in the distal or unilateral pulmonary artery, are used to visualized the pulmonary vasculature. Distal stenotic lesions, whether congenital or acquired and owing to the perils of recanalization of thrombotic lesions, should be carefully balloon-dilated. An aortogram should be performed to exclude aortopulmonary connections.
Exercise testing
The exercise capacity may reflect the disease severity and also assist in the the prognostic evaluation for PAH patients. For patients with Eisenmenger syndrome, exercise testing also provides information on the change in arterial oxygen saturation during exercise. The exercise capacity is assessed by either measurement of a 6-minute walk test distance or by cardiopulmonary exercise testing with measuring the peak oxygen consumption. A reduced peak oxygen consumption and a reduced 6-minute walk distance are associated with an impaired prognosis for PAH patients. For cyanotic patients, early termination of exercise may be related to an increase in right-to-left shunting and arterial hypoxemia rather than to ominous pathophysiological abnormalities, including poor ventricular function, autonomic nervous dysfunction or myocardial ischemia. In addition, chronic cyanosis represents multiorgan disease and this is associated with abnormalities such as systemic endothelial dysfunction, and these factors may confound the prognostic value of determining the exercise capacity of Eisenmenger patients. Currently, the 6-minute walk test represents the preferred method to assess the exercise capacity and appraise the therapeutic effects in PAH patients. The 6-minute walk test is robust and it is currently is the only exercise test modality approved by the Food and Drug Administration as an end point for prospective clinical trials in this setting.
High-Resolution or multi-detector computed tomography with 3 dimensional reconstruction
High-resolution or multi-detector computed tomography with 3 dimensional reconstruction computed tomography is especially useful to assess for pulmonary arterial thrombi and to exclude intrapulmonary hemorrhage or infarction in PAH patients PAH. It is also the imaging modality of choice for assessing the lung parenchyma.
Management
Early diagnosis and treatment for a large systemic-to-pulmonary shunt is critical for preventing Eisenmenger syndrome. The treatment options for patients with PAH associated with congenital heart disease are limited to palliative measures and heart-lung transplantation for highly selected patients with Eisenmenger syndrome. The treatment may include the use of supplemental oxygen, digitalis, diuretics, vasodilators and anticoagulants, or lung transplantation and repair of the congenital heart defect(s), or heart-lung transplantation.11)12)24-29)
Medical Management
There is no cure for PAH, but the medical treatment has recently improved dramatically, offering both relief from symptoms and prolonged survival. The mainstays of current medical therapy fall into several classes, including vasodilators, anticoagulants, antiplatelet agents, antiinflammatory therapies and vascularremodeling therapies. Many of the most effective agents have pleiotropic effects. Epoprostenol is a vasodilator, a platelet inhibitor and an antiinflammatory agent, whereas the endothelin-receptor antagonist bosentan is a vasodilator, an antiinflammatory agent and a remodeling mediator.
General Management
Patients with Eisenmenger syndrome must avoid circumstances that may exacerbate their pulmonary vascular disease.
Exercise should be guided by the patients' symptoms with strict limits adhered to. Avoidance of travel to high altitude, although air travel is permitted, should be advised because of the potential for alveolar hypoxia leading to further pulmonary arterial vasoconstriction and worsened symptoms.
Pregnancy, oral contraceptives and hormone replacement therapies should be avoided because of the increased risk of thromboembolic events. Pregnancy can be fatal for patients with PAH both during the course of delivery and postpartum.
Conservative Therapy
Digitalis and diuretics
The efficacy of inotropic agents for right heart failure remains controversial. Diuretics may be useful for patients with Eisenmenger syndrome and severe right heart failure to relieve hepatic congestion or to decrease the intravascular volume. This must be done cautiously for those patients who have significant polycythemia, and physicians needs to weigh the danger of the increased hyperviscosity, which can lead to stroke and related complications.
Anticoagulation
Patients with Eisenmenger syndrome are at risk for thromboembolic events that can happen due to a sedentary lifestyle, right heart failure and sluggish pulmonary blood flow. Even a small pulmonary embolus can be life threatening in patients with pulmonary vascular disease, and this can cause sudden death. Although there have been no adequate studies to demonstrate the efficacy of anticoagulation for patients with Eisenmenger syndrome, the risk of bleeding and particularly hemoptysis warrant consideration. When warfarin is used, the aim is to maintain the international normalized ratio(INR) at 1.5 to 2.0 for most patients.
Phlebotomy
Many patients with Eisenmenger's syndrome tend to stabilize the hematocrit in the 60% to 65% range for many years or even decades, and they no longer require phlebotomy. When a hematocrit reaches the 65% to 70% range or if the patient is symptomatic with a lower hematocrit, then exchange transfusion with plasma or crystalloid is indicated and this needs to be done carefully because simply removing blood can decrease the systemic vascular resistance and result in a sudden hypoxic event. Supplemental iron therapy is also indicated for iron deficiency even if the patients are polycythemic.
Vasodilator Therapy
Pulmonary vasodilator therapy has been used and this is based on the treatment of primary pulmonary hypertension(PPH). The main concept is that pulmonary vasoconstriction plays a role in the development of the pulmonary vascular disease. Although the mechanisms may be different for patients with Eisenmenger syndrome compared with PPH patients, there is now evidence that patients with Eisenmenger syndrome may also benefit from vasodilator and/or antiproliferative therapies such as intravenous epoprostenol and Bosentan, which were previously reserved for PPH patients.
Before initiation of pulmonary vasodilator therapy, patients should undergo cardiac catheterization: (1) to assess the "operability" and (2) for acute pulmonary vasodilator testing in those patients who are considered inoperable, i.e., Eisenmenger syndrome as above. If a patient responds to acute vasodilator testing with a decrease in the pulmonary vascular resistance of ≥30% after the administration of inhaled nitric oxide (10-80 ppm) or intravenous epoprostenol and a similar response is demonstrated to acute testing with sublingual CCB, then they can be offered treatment with long-term oral CCBs if they are not in significant right heart failure. This is based on previous studies for patients with PPH who demonstrate acute pulmonary vasoreactivity and subsequent clinical and hemodynamic improvement as well as increased survival with long term administration of oral CCBs. Yet a significantly lower acute response rate was demonstrated for patients with PAH associated with congenital heart defects as compared to that for patients with PPH. In a small study, only 7% of the 94 patients with pulmonary hypertension associated with congenital heart defects responded to acute vasodilator testing.
Prostacyclin
As a potent vasodilator, prostacyclin has been shown to improve the hemodynamic function, the exercise tolerance and the quality of life. The drug comes in 3 forms: intravenous prostacyclin(epoprostenol 0.5-1 ng/kg at starting dose, but currently unavailable in Korea), inhaled prostacyclin(iloprost), and oral prostacyclin(beraprost). There is a risk for rebound PAH with abrupt discontinuation of therapy.
Type 5 phosphodiesterase inhibitors
Sildenafil is a selective inhibitor of type 5 phosphodiesterase inhibitors, which break down cGMP and limits cGMP-mediated NO vasodilation. Because Sildenafil can prolong the activity of NO, it has been used to prevent the rebound PAH even with discontinuation of inhaled NO therapy(0.3 mg/kg/ day -20 mg/day).
Endothelin receptor antagonist
Endothelin 1, a potent vasoconstrictor, achieves its activity though two types of endothelin receptors, ETA and ETB. Bosentan, the dual-receptor antagonist(now available in Korea), has been shown to improved the functional capacity and hemodynamics. According to The Bosentan Randomized Trial of Endothelin Antagonist Therapy-5(BREATHE-5), which was recently reported in 2006, Bosentan did not worsen oxygen saturation, it reduced the pulmonary vascular resistance index, with a decrease of the mean PAP, and the exercise capacity was increased in the patients with World Health Organization functional class III Eisenmenger syndrome. Bosentan was well tolerated and it improved the exercise capacity and hemodynamics without compromising the peripheral oxygen saturation.32)
Interventional/Surgical Treatment
The two options are atrial septostomy and lung or heart-lung transplantation.
Atrial septostomy
Patients with PPH and in whom the foramen ovale(PFO) is patent live longer than those without a patent FO. Furthermore, patients with Eisenmenger syndrome that's caused by an atrial septal defect(ASD) have a better prognosis than PPH patients who have an intact atrial septum. However, pulmonary vascular disease may occur in up to 5% to 10% of patients with untreated ASDs, and this is predominant in females. An atrial septostomy may improve symptoms and survival for postoperative patients with advanced pulmonary vascular disease and right heart failure and who have no significant residual communication between the left and right circulation.
Transplantation
While successful heart-lung transplantation, as well as lung transplantation with repair of the congenital heart defects, might be available, there are several limitations to these procedures. Patients with Eisenmenger syndrome have the highest perioperative mortality and the lowest 1-month survival rates among all the lung transplant recipients. For patients with Eisenmenger's syndrome, the 1-year and 5-year survival rates after lung transplantation are 52% and 39%, respectively.
The Risk Factors for Sudden Cardiac Death
The cause of death for the majority of the follow-up studies on Eisenmenger syndrome has displayed two distinct patterns. One set of patients appears to develop progressive heart failure, which is the cause of death, whereas a second set dies suddenly. The patients dying from heart failure appears to be increasing in comparison with sudden death in the recent studies.
Hemoptysis, which was the cause of death in an earlier era, remains a common cause of clinical morbidity, but not of death. The third important observation is that patients with Eisenmenger syndrome associated with complex heart disease die sooner than those with simple lesions, and it can be anticipated that the cardiac function may be relatively well preserved for longer periods in patients with simple lesions.
For the mechanisms of sudden death in young patients, either those with normal hearts or those with previous operations for congenital heart disease, acute arrhythmia is increasingly being recognized to be the terminal event. Ventricular tachycardia or fibrillation was noted in the patients for whom an electrocardiogram was recorded during the terminal event. It would be reasonable to suspect that sudden deaths in the Eisenmenger population could be arrhythmic in origin. Supraventricular or ventricular arrhythmia may produce acute worsening of the cardiac performance and CO, and so lead to death.
The risk factors associated with a failing heart are currently better understood. A higher NYHA functional class and the clinical and laboratory parameters of heart failure are predictors of mortality. The age at clinical presentation is also a risk factor and it may be presumed that younger patients have more severe disease or rapid progression. The degree of hypoxemia is also correlated with the NYHA functional class and it is associated with a poorer prognosis, as is an elevated right atrial mean pressure, which probably reflects ventricular failure.
Sustained arrhythmias, be they supraventricular or ventricular, have been documented on routine electrocardiograms or 24-h Holter monitoring, and previously documented arrhythmia was indeed a marker for death. Some symptoms like palpitations and syncope appeared to be relatively common in the Mustard/Senning study, and they were predictive of sudden death. Syncope was associated with a poor outcome. Alterations in the baseline electrocardiogram, which may reflect abnormal depolarization or repolarization, could be predictors of death. It is clear that patients with Eisenmenger syndrome should be aggressively monitored for diagnosing potentially lethal arrhythmias and any symptoms of arrhythmia warrant careful follow-up with serial long-term Holter recordings. In patients with documented sustained arrhythmia, aggressive therapy with antiarrhythmia medications, pacing or even an implantable defibrillator may be considered. Selective pulmonary vasodilator therapy can be done for patients with increasing heart failure consequent to the progression of pulmonary vascular disease, as there is some evidence that this is also beneficial in the Eisenmenger population. For patients with suspected or documented arrhythmia, arrhythmia prevention strategies should take precedence, if achievable, and this would be of obvious benefit.30-34)
Predictable Length of Survival
Some studies34) have shown a predicted median survival of 52.6 years for adult patients with the Eisenmenger syndrome. These patients may survive into their third or fourth decade if managed appropriately. The survival rates at 30, 40, and 55 years of age were 75%, 70%, and 55%, respectively. Compared with PPH, survival is clearly better for PAH patients with congenital heart disease. PPH represents a life-threatening disease with a devastating prognosis and a reported median survival of 2.8 years. Several potential mechanisms account for this superior prospect of survival compared with PPH. First, in PAH patients with congenital heart disease, the right ventricle is subjected to high pressures from birth or from infancy, and so it may be better trained to support the systemic pulmonary pressures, which can reduce the incidence of early right ventricular failure. Second, in patients with PPH, the pulmonary hypertension itself limits the pulmonary blood flow and thereby it reduces the systemic blood flow during exercise. Patients with PAH-associated shunts maintain or increase their systemic cardiac output during exercise via right to left shunting.25)34)
Conclusion
PAH is, to a variable degree, associated with congenital heart disease, and this depends on the size and location of the underlying cardiac defect, as well as on the repair status. Eisenmenger syndrome is at the extreme end of PAH that presents with or without reversed shunting and associated cyanosis. The prospects for survival have improved for patients with Eisenmenger syndrome in the current era. The great advances in operative skill and medical treatment, such as the new vasodilators, allow young patients with congenital heart defects to undergo early diagnosis and early corrective surgery. Eisenmenger syndrome has previously implied irreversibility and inoperability for a subgroup of patients with PAH and congenital heart defects. Today, although there have been many advances in understanding the pathophysiology of Eisenmenger syndrome and also advances in treating this disease, there is still no cure for this progressive condition once it is established, aside from lung or heart-lung transplantation, which is associated with significant morbidity and mortality. The newly developed vasodilators are well tolerated and they improve the exercise capacity and hemodynamics without compromising the peripheral oxygen saturation in patients with Eisenmenger syndrome.




 XML Download
XML Download