

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Extragonadal germ cell tumors (GCTs) are thought to derive from primordial germ cell precursors that become sequestered during embryologic migration and survive in ectopic locations1,2. Their anatomic location can vary widely, with tumors occurring along the body midline, following the course of embryonal germ cells. Preferred sites include the mediastinum, the sacrococcygeal and pineal regions, the neck, and the retroperitoneum3-6. Intraabdominally located extragonadal GCTs are extremely rare, with only a few cases reported in the English literature1-3,5-10. We describe here an 18-month-old girl with an intraabdominal mixed GCT and liver metastasis, who was treated by surgical excision and postoperative chemotherapy.
CASE REPORT
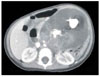
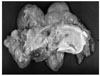
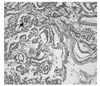
An 18-month-old girl was transferred to our hospital due to a large left upper abdominal mass, found incidentally while bathing. She had no associated complaints. On physical examination, an approximately 11 cm well-defined, firm, movable globular mass was palpated in the left upper quadrant of her abdomen. Her serum α-fetoprotein (AFP) concentration was elevated to 950 ng/mL (normal, < 8 ng/mL), whereas other biochemical parameters and tumor markers, including carbohydrate antigen (CA) 125 and β-human chorionic gonadotropin (β-hCG) were within normal limits. Abdominal ultrasonography (USG) showed a huge cystic mass with multiseptation, calcification, and solid components in the left upper quadrant of the abdomen. Abdominal computed tomography (CT) scanning revealed a 12 × 8 cm, well demarcated, cystic mass with calcification and solid and fat components in the left upper quadrant of the patient's abdomen, along with a 1 cm, well-defined, ovoid, low density nodule in segment IV of her liver (Fig 1). An explorative laparotomy revealed a 9 × 8 cm, globular encapsulated mass, located between the greater omentum and transverse mesocolon, and a 1 cm, ovoid nodule in segment IV of the liver. The mass showed no evidence of invasion of adjacent organs. Both ovaries were normal at the time of surgery. The mass was completely excised surgically, and the hepatic lesion underwent wedge resection. Grossly, the tumor was a 10 × 9 cm, whitish to gray, globular solid mass (Fig. 2); sectioning revealed semisolid grayish material and cartilaginous tissue. Pathologic examination revealed a mixed malignant GCT made of two components, a mainly endodermal sinus tumor and a small mature teratoma. We also observed a loose reticular pattern and rounded papillary processes with central capillary, Schiller-Duval bodies. The tumor was immunohistochemically positive for AFP. The resected hepatic specimen was a metastasis of the mixed GCT and was histologically identical to that component of the primary tumor (Fig 3). Post-operatively, the patient received five courses of BEP chemotherapy, consisting of 15mg/m2 of bleomycin, 100mg/m2 of etoposide, and 40mg/m2 of cisplatin. Serum AFP concentration returned to normal 3 months after the operation. The patient was followed up annually with measurement of serum AFP concentration and abdominal CT scans. At present, more than 10 years after surgery, this patient is alive and well, with no evidence of tumor recurrence.
DISCUSSION
Extragonadal GCTs are defined as germ cell tumors displaying a histology associated with gonadal origin, but located outside the gonads. Extragonadal GCTs are relatively uncommon, accounting for fewer than 5% of all GCTs2. Their pathologic subtypes vary widely and include mature and immature teratomas, seminomas, yolk sac tumors, embryonal carcinomas, choriocarcinomas, and mixed GCTs. Mixed GCTs, which are rare, include components of at least two malignant GCTs.
GCTs are thought to originate from primordial germ cells, which migrate to the primitive gonadal glands in the urogenital ridge1. The histogenesis of extragonadal GCTs, however, remains unclear, although several hypotheses have been proposed based on embryologic and histopathologic considerations10. The first hypothesis states that extragonadal GCTs derive from the aberrant differentiation of somatic cells. A more persuasive hypothesis states that extragonadal GCTs develop from primordial germ cell precursors, with the latter being misplaced or arrested during their embryologic migration to the gonads, resulting in their survival in ectopic locations. According to this hypothesis, the tumor observed in our patient may derive from the arrest of germ cell migration through the bowel wall, mesentery or omentum. The third hypothesis states that metastasis from the occult focus of testicular or ovarian gonadal GCTs may give rise to extragonadal GCTs; however, both ovaries were normal in our patient, and thorough examination has excluded this possibility.
The anatomic locations of extragonadal GCTs vary widely. These tumors have been found along the median line of the human body, which derives from the embryonic germ cell ridge. Preferred sites include the mediastinum, the sacrococcygeal and pineal regions, the neck, the intracranial region, the retroperitoneum, the liver, and the pelvis. Although pelvic and/or retroperitoneal extension of a sacrococcygeal tumor is not unusual, an exclusively retroperitoneal or abdominal location is uncommon. Intraabdominally located extragonadal GCTs are extremely rare, with only 9 pediatric patients with these tumors have been reported in the English-language literature before the present report (Table 1), and long-term follow up of these patients was lacking. Due to the large size of the tumor in our patient, a precise site of origin could not be determined. Nevertheless, our case appears to be extraordinary due to the rare location of the primary tumor, the presence of a metastatic site in the liver at the time of surgery, and the long-term survival of our patient.
Extragonadal GCTs have a female preponderance. Most tumors are diagnosed at an advanced stage and are locally unresectable with metastases to the lungs or liver2. The advanced stage at diagnosis is possibly due to the retroperitoneal location of these tumors and the lack of obstructive symptoms, despite large tumor size.
Most intraabdominal and retroperitoneal extragonadal GCTs derive from metastases, usually from primary testicular and ovarian tumors. It is difficult to distinguish morphologically between primary and metastatic GCTs, making clinical correlation decisive in these patients.
Mixed GCTs secrete AFP and/or β-hCG, depending on the components of the tumor. In our patient, serum AFP, a hallmark of endodermal sinus tumors, was elevated.
Management of mixed GCTs is geared toward the most malignant components. The preferred treatment for children with endodermal sinus tumor is complete surgical resection, with adjuvant chemotherapy evaluated on an individual basis. Surgical resection is determined by the extent of disease at the time of diagnosis. In most patients, however, it is difficult to remove the lesion completely due to its diffuse bulky extension. In these patients, a limited biopsy for tissue diagnosis, followed by chemotherapy and resection of the residual mass, has been recommended. Adjuvant chemotherapy after surgery has increased the survival rate of patients with GCTs10. Although several chemother-apeutic regimens have been employed, the best results were achieved using BEP, consisting of bleomycin, etoposide, and either standard or high dose cisplatin.
Although the rarity of extragondal GCTs make it difficult to predict the long-term outcomes in patients with these tumors, children with malignant germ cell tumors arising in the abdomen and retroperitoneum, including our patient, have an excellent prognosis, despite the advanced stage of disease at diagnosis2.
Our findings suggest that surgical resection and aggressive combination chemotherapy could improve the survival of pediatric patients with extragonadal mixed GCTs.

 XML Download
XML Download