

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
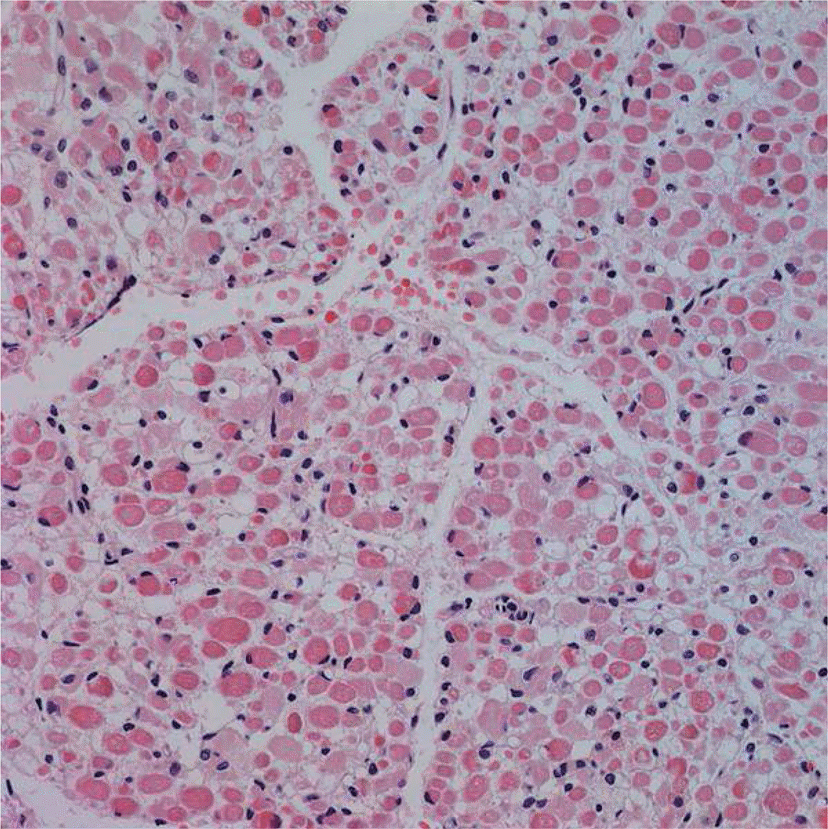
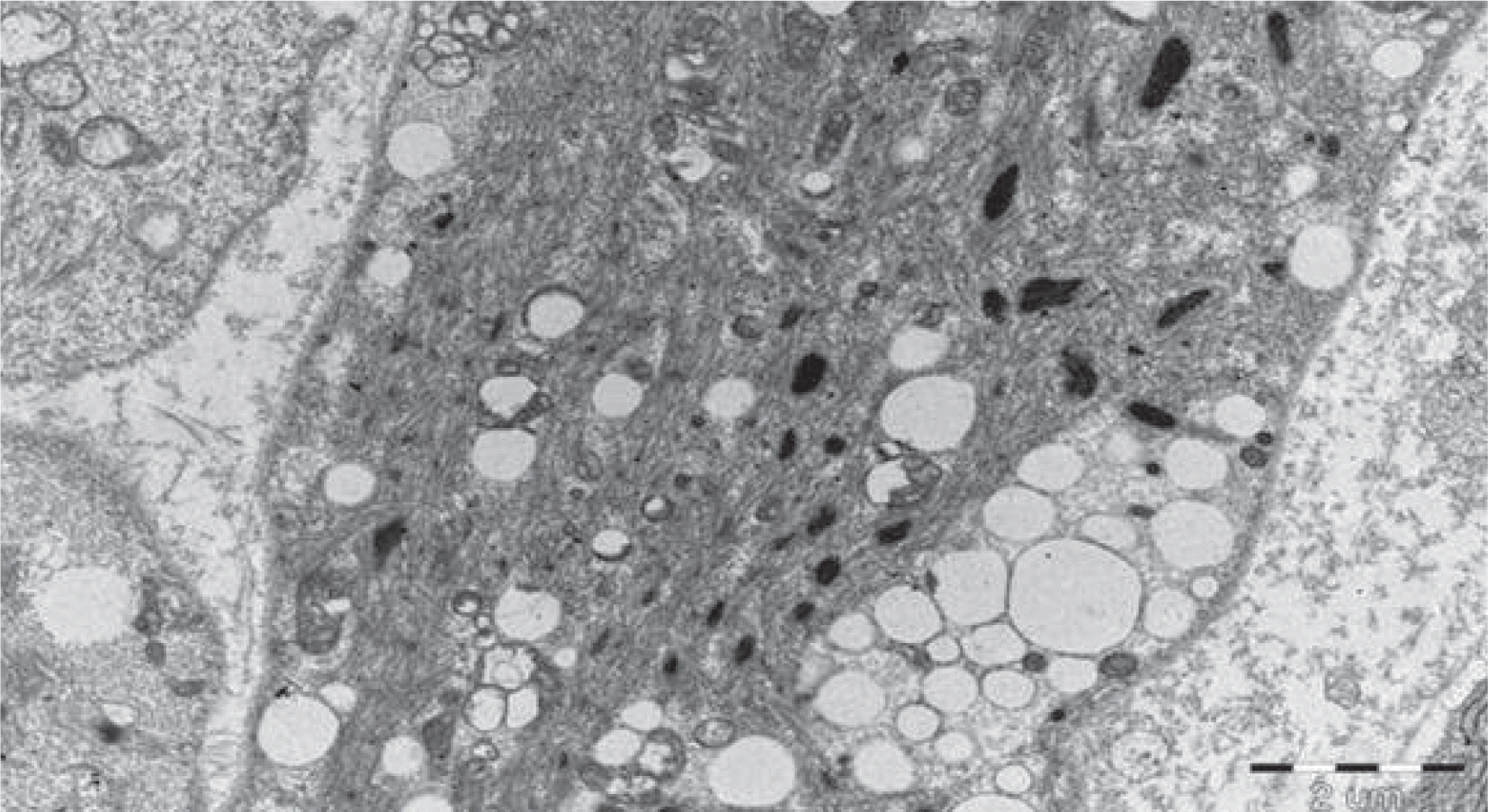
Glycogen storage disease (GSD) is a group of heterogeneous disorders of glycogen metabolism that results in abnormal storage of glycogen in multiple organs. Clinical manifestations of GSD vary according to the basic enzyme defect. Only types II, IV, V or VII of GSD have been known to manifest in the infantile period. Of the 11 types of GSD, the congenital subtype of GSD type IV is characterized by severe neonatal hypotonia, multiple contractures, polyhydramnios, and fetal hydrops. We report a case of a patient born at a gestational age of 34 weeks and 3 days with fetal hydrops, joint contractures, and akinesia. Muscle biopsy results were highly indicative of GSD. This is the first case of suspected GSD in Korea presenting as fetal hydrops. The possibility of other disorders associated with glycogen metabolism should be considered in fatal fetal hydrops patients with severe hypotonia and arthrogryposis, and aggressive investigations such as muscle biopsy should be performed for early diagnosis.
REFERENCES
1). Hicks J., Wartchow E., Mierau G. Glycogen storage diseases: a brief review and update on clinical features, genetic abnormalities, pathologic features, and treatment. Ultrastruct Pathol. 2011. 35:183–96.

2). Moses SW., Parvari R. The variable presentations of glycogen storage disease type IV: a review of clinical, enzymatic and molecular studies. Curr Mol Med. 2002. 2:177–88.
3). Alegria A., Martins E., Dias M., Cunha A., Cardoso M., Maire I. Glycogen storage disease type IV presenting as hydrops fetalis. J Inherit Metab Dis. 1999. 22:330–2.
4). Maruyama K., Suzuki T., Koizumi T., Sugie H., Fukuda T., Ito M, et al. Congenital form of glycogen storage disease type IV: a case report and a review of the literature. Pediatrics interna- tional. 2004. 46:474–7.
5). Raju GP., Li H-C., Bali DS., Chen Y-T., Urion DK., Lidov HG, et al. A case of congenital glycogen storage disease type IV with a novel GBE1 mutation. J Child Neurol. 2008. 23:349–52.
6). Cox PM., Brueton LA., Murphy KW., Worthington VC., Bjelogrlic P., Lazda EJ, et al. Early-onset fetal hydrops and muscle degeneration in siblings due to a novel variant of type IV glycogenosis. Am J Med Gen. 1999. 86:187–93.
7). L'herminé-Coulomb A., Beuzen F., Bouvier R., Rolland M., Froissart R., Menez F, et al. Fetal type IV glycogen storage disease: clinical, enzymatic, and genetic data of a pure muscular form with variable and early antenatal manifestations in the same family. Am J Med Genet A. 2005. 139:118–22.
8). Lee Y-C., Chang C-J., Bali D., Chen Y-T., Yan Y-T. Glycogen-branching enzyme deficiency leads to abnormal cardiac development: novel insights into glycogen storage disease IV. Hum Mol Genet. 2011. 20:455–65.
9). Konstantinidou A., Anninos H., Dertinger S., Nonni A., Petersen M., Karadimas C, et al. Placental involvement in glycogen storage disease type IV. Placenta. 2008. 29:378–81.
 XML Download
XML Download