

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Adrenomedullin (AM) is a 52-amino-acid peptide that is formed from the proteolytic cleavage of pro AM. AM belongs to the calcitonin superfamily on account of its structural similarity to the calcitonin gene-related peptide (CGRP) and amylin4). Since its isolation from an adrenal tumor (pheochromocytoma) in 1993, AM has been shown to be present in many mammalian tissues and fluids, regulating a variety of physiological activities in normal and malignant settings3). The pleiotopic activities of AM were recognized within a few years of its initial characterization as a vasodilatory peptide3,4).
The expression and secretion of AM has been demonstrated in many tumors and biological fluids and has been suggested to modulate a variety of physiological processes3). AM expression is stimulated by cytokines and hypoxia and its promoter contains several putative hypoxia response elements16). Hypoxia induces AM expression and secretion in many cell types including carcinoma cell lines and endothelial cells10). Moreover, hypoxia is a common feature of the microenvironment in solid tumors and is one of the driving forces for cancer growth and progression. AM has been suggested to play a role as a promoter of these processes2). AM promotes the formation of tumors by stimulating the autocrine growth and survival of tumor cells, as well as through its paracrine effects on the surrounding vessels. The possible intracellular mechanisms underlying the effects of AM suggest its potential role in tumor progression. AM signal transduction differs according to the cell types and involves several pathways3,11). Increasing evidence suggests that the activation or disruption of AM signaling may contribute to various pathologies, such as, ischemia-induced damage and neoplastic growth.
Cell growth, division, differentiation and deaths are now known to be regulated in part by the mitogen activated protein kinase (MAPK) pathway9,14). Cellular signal transduction is a two step-process: a signaling molecule is sensed by a receptor on the target cell and the receptor is then activated. The response is amplified when the receptor sensing the signal is a catalyst, a kinase for example. At least three parallel MAPK pathways have been identified, extracellular signal regulated kinase (ERK), stress activated protein kinase (SAPK), also known as JNK for c-jun-N-terminal kinase), and the p38/MAPK pathways.
It is becoming increasingly evident that AM plays a role in tumor proliferation. Therefore, this study examined the cell proliferation stimulated by AM as well as the the signaling pathways responsible for the activations of ERK 1 and 2 by AM in human osteosarcoma was warranted.
MATERIALS AND METHODS
1. Cell culture
The human osteosarcoma (OS) cell lines MG 63 and U2OS (Korean Cell Line Bank, Seoul, Korea) were cultured. All experiments were carried out in Eagle's modified minimal essential medium containing 10% inactivated fetal bovine serum (GIBCO, Carlsbad, CA). In all the experiments, the OS cells were cultured in 10% serum and harvested with trypsin which was blocked by complete media as soon as the cells had become detached. Trypan blue exclusion was used to test for the cell viability in order to exclude the possibility that the cells had been damaged by trypsin. In all the experiments, the cells were left to recover from trypsinization for 24h before being treated. The media were replaced after the third day, only in the experiments requiring longer treatment times. In all experiments, the cells were plated at the same density, i,e., at densities equivalent to 3,000 cells/well of a 96 well plate.
2. Chemicals
AM (1-52) was purchased from Sigma (St. Louis, MO) and was used at concentrations of 10 nM, 50 nM, and 100 nM in both cell lines.
3. Cell proliferation assay by cell count
The level of cell proliferation was determined by counting the cells, as described elsewhere1). Briefly, the MG63 and U2OS cells (10,000 cells/well) were plated in a 12-well tissue culture plate containing the minimum essential medium (MEM) with 10% of fetal bovine serum (FBS). Immediately after attachment to the plates, cells were switched to serum-free MEM for 24h. Subsequently, the cells were either left untreated or treated with AM (1, 10, or 100 ng/ml) in the presence or absence of PD98059 (10 uM) and incubated at 37℃ for 3 days. Subsequently, they were trypsinized and the living cells (trypan blue exclusion) were counted using a hemocytometer.
4. Cell proliferation measurements using an enzyme-derived formazan dye from the tetrazolium salt (WST-1) assay
The viabilities of the osteosarcoma cells was determined using a WST-1 quantitative colorimetric assay. The assay detects living cells only and the results are thus dependant on the degree of cellular viability. In this study, the sulfonated tetrazolium salt,4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1, 3-benzene disulfonate (WST-1) assay, was used because this compound produces a highly water-soluble formazan dye, which makes the assay procedure easier to perform. Subconfluent osteosarcoma cells grown in 96-well plates were treated with the agents for 48h. WST-1 (16 mg) and 0.2 mM 1-methyl-phenazium methylsulfate were dissolved in a 20 mM 4-(2-hydroxyethyl)-1-piperazine ethanesulphonic acid (HEPES) buffer (pH 7.4). Ten ul and 10 ul of the reaction solution was added immediately to 100 ul of the culture medium per well. The cells were incubated for an additional 2 h at 37℃ in a humidified atmosphere of 5 % CO2 and 95 % air, and the cell viability was determined using a WST-1 assay. The absorbance of the lysate was measured at 405 nm for the test samples and 650 nm as a reference wavelength using an Immuno Mini NJ-2300 microplate reader (Japan Intermed, Tokyo, Japan).
5. MAP kinase activity
The antibodies (Promega, Madison, WI, USA) specific to the phosphorylated, active forms of extracellular signal-regulated kinase-1 and -2 (ERK1/2) were used for the Western blotting. After culturing in 60 mm dishes, the cells were collected and added to 600 ul extraction buffer (25 mM HEPES, 0.5% Triton X-100, 5 mM ethylene glycol-bis (β-aminoethyl ether)-N, N, N', N'-tetraacetic acid (EGTA), 5 mM ethylenediaminetetraacetic acid (EDTA), 1 mM phenylmethysulfonyl fluoride, 50 nM beta-glycerol phosphate, and 10 mM sodium orthovanadate). The samples were then heated to 95℃ for 5 min, sonicated and centrifuged for 10 min at 15,000×g. Aliquots of 20 ul were mixed with 5 ul of a 5x sample buffer and subjected to SDS-polyacrylamide gel electrophoresis using 10% gels. The proteins were blotted onto Immobilin-P transfer membranes (Millipore, San Jose, CA, USA) and the nonspecific binding was blocked using Tris-buffered saline (TBS) containing 5% skim milk for 90 min at 37℃. The membranes were then incubated with the primary antibody for 3 h at room temperature and the protein bands were revealed using horseradish peroxidase-conjugated goat antirabbit immunoglobulins and visualized using an enhanced chemiluminescence detection system. The activated kinase bands were quantitated by densitometry using NIH Image software (National Institutes of Health, Bethesda, MD) soft ware analysis.
6. MAP kinase inhibition studies
The MG63 and U2OS cells were incubated for 30 min in the presence of 10 uM of 2'-amino-3'-methoxyflavone (PD 98059; a specific MEK1 inhibitor) (Calbiochem, CA, USA). The cells were also treated with the MAPK inhibitors and cultured in the absence of AM for comparison purposes. The number of viable cells was determined by Western blotting, as described above.
RESULTS
1. AM treatment increases cell survival in osteosarcoma cell lines
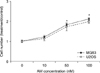
The proliferative effects of AM on the two osteosarcoma cell lines (MG63, U2OS) were assessed by cell counting. Treatment with AM for 24 h, in cultures grown in medium containing 10% inactivation. FBS induced dose-dependant increases in the numbers of cells in both cell lines (Fig. 1) and significant increases were observed at AM concentrations of 50 nM or greater. The stimulations were maintained for 48 h (Fig. 2).
The cell viability was determined using WST-1 assays. Fig. 3 shows that AM increased the cell viability in both cell lines in a dose-dependant manner up to 50 nM. AM at 50 nM increased the cell viability of MG63 and U2OS cells to approximately 285±20.5% and of U2OS cells 262± 15.3% of the control level, respectively. Treatment with AM for 24 h produced a dose-dependant increase in WST-1 assay (Fig. 4). The stimulation were maintained for 48 h.
2. Effect of PD98059 on AM-stimulated osteosarcoma cell lines
As shown in Fig. 1, 100 nM of AM caused the maximum proliferation of both cell lines in the serum-deprived media. PD98059, selective inhibitor of MEK, was used to determine the role of ERK activation in mitogenesis. As shown in Fig. 5, treatment with 10 uM PD98059 completely inhibited AM-induced proliferation in both cell lines.
3. AM treatment increases expression of ERK MAP kinase
Polyclonal antibodies against the conserved, non-phosphorylated regions in mammalian ERK1 and ERK2 MAP kinase were used to probe the immunoblots of the osteosarcoma cell lines (MG63, U2OS). Phospho-specific antibodies, which detect the active forms of the MAP kinase, were used. ERK1 and 2, MAPK were found to be activated within 3 hours after the AM treatment. A strongly immunoreactive protein of molecular weight of about 42 kDa was obtained. Treatment with AM at 100 nM caused a two-fold increase in the phosphor-MAP kinase levels without affecting the total MAPK protein levels (Fig. 6). Furthermore, this increase in the phosphor-MAP kinase level was prevented by a co-treatment with 100 nM AM and 10 uM (Fig. 7).
DISCUSSION
This study examined the effects of AM, as a mediator of cancer proliferation, on osteosarcoma cell lines. It was found that AM stimulated the proliferations of the MG63 and U2OS osteosarcoma cell lines. Treatment with AM produced a dose-dependant increase in the numbers of actively growing osteosarcoma cell lines. This effect was significant at AM concentrations of 50 nM or greater. Futhermore, this stimulation was maintained for 48 h. AM inhibited MAPK and stimulated MAPK phosphatase, which may be related to the mitogenic activity of AM. Moreover, the AM-induced cell proliferation of the AM-stimulated osteosarcoma cell lines (as determined by cell counting) was also blocked by PD98059 (a MEK inhibitor). These findings prompted us to study the signal transduction pathway of MAPK activation inactivate by AM.
Since the seminal report by Kitamura et al.4), a number of studies have reported AM expression in different tumor cell lines and malignant tissues. For example, AM is expressed in brain tumors13) and binds to a neuroblastoma cell line15). Martinez et al.8) demonstrated AM expression in the airway epithelium as well as in several lung cancer cell lines. Polyclonal antibodies specific to AM can block the binding of AM to its cellular receptors and significantly reduce U87 glioblastoma cell proliferation in vitro. Furthermore, the intratumoral administration of the anti-AM antibody resulted in a dramatic decrease in subcutaneous U87 xenograft tumors12).
Mammalian MAPKs consist of three major subfamilies, ie., ERK MAPKs, c-Jun NH 2 -terminal kinase (JNK) MAPKs/SAPKs, and the p38 MAPKs. Each MAPK has its specific substrates and functions, which range from the regulation of cell proliferation to the cell death. One of the most important pathways for cell proliferation involves the ERK MAPKs. ERK MAP kinases are located centrally in the mitogenic signal cascades, which is a series of phosphorylation reactions involving the cell surface receptors, Ras, Raf, and MEK or protein kinase c (PKC). The ERK MAP kinase pathway, which contains several proto-oncogenes and factors, has been studied extensively in a variety of human cancers. The MAP pathways are known to be primarily responsible for transducing and amplifying extracellular signals intracellulary, which leads to adaptation to the environmental changes. On the other hand, the ERK 1/2 MAPK pathway is induced by several growth factors and mitogens, which controls cell proliferation. I contrast, the JNK1- and p38-involved pathways are activated by many proinflammatory cytokines and by environmental stress, which lead to alter gene expressions and apoptosis. This study suggests that the AM activated MAPK pathways underlie osteosarcoma cell proliferation.
ERK is involved in the proliferative response of osteoblasts to a variety of mitogens. Osteoblasts, once they have completed their bone-forming function, are either entrapped in the bone matrix or become lining cells. ERK-1 and ERK-2 are also important in osteoblastic cell proliferation and differentiation5,7). Moreover, the MG63 and U2OS cell lines are widely used as osteoblastic models because they retain many osteoblastic properties including high alkaline phosphatase activity and 1α,25-dihydroxyvitamin D3 induced fibronectin, osteocalcin, and interlukin-66).
Western blotting showed dose dependant increases in ERK MAP kinase activity in the AM-treated MG63 and U2OS cells. Moreover, the PD98059 treatment, which blocks the ERK MAP kinase cascade, confirmed that AM induced proliferation through ERK MAP kinase cascade. These results suggest that AM plays an important role in MG63 and U2OS proliferation and the AM-induced ERK kinase pathway transduces the signals in these cell lines.
In conclusion, AM stimulates the activity of ERK in the two osteosarcoma cell lines examined, which is blocked by PD98059. The inhibition of ERK by PD98059 suggests that the mitogenic effect of AM is mediated in part through ERK dependant activation. Therefore, AM inhibition offers a therapeutic strategy for preventing osteosarcoma progession. However, further studies will be needed to exam the expression of AM and its distribution in human osteosarcoma tissues.






 XML Download
XML Download