

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Lineage switch in relapsing leukemia is a rare event. The rate of conversion from ALL to AML has been found to be 6-9%, and most of the lineage switches have been reported in children [1-5]. In contrast, conversion from AML to ALL is extremely rare in children [6-11]; further, only 3 such cases have been reported in adults [12-14]. To our knowledge, no cases of conversion from erythroleukemia to ALL have been reported to date. In cases showing relapse with lineage switch, most leukemic clones have a different morphology, phenotypic lineage, and distinct molecular characteristics. In general, the time between initial diagnosis and relapse with lineage switch is 1-4 yr [1]. We describe the case of an adult patient with erythroleukemia who showed ALL relapse only 4 months after initial diagnosis; the relapsed ALL presented with cells showing a different morphological and phenotypic lineage but with the same clones and some clonal evolution.
Go to : 
CASE REPORT
A 62-yr-old male patient presented with dyspnea and pancytopenia. Neither splenomegaly nor lymphadenopathy was found. He had no previous medical history. Peripheral blood analysis yielded the following findings: hemoglobin, 5.6 g/dL; platelet count, 126×109/L; and leukocyte count, 2.2×109/L, with 88% lymphocytes, 6% segmented neutrophils, 2% monocytes, 1% myelocytes, 1% eosinophils, 1% basophils, and 1% atypical lymphocytes.
Bone marrow (BM) analysis revealed that 52% of the nucleated cells were erythroid precursors and that 39.2% of the non-erythroid cells were blasts with 2 types of morphology. The predominant blasts were small- to medium-sized with dispersed nuclear chromatin, 2 or 3 prominent nucleoli, and a moderate amount of cytoplasm, with a few blasts containing cytoplasmic vacuoles (Fig. 1A). None of the hematopoietic cell lineages showed dysplastic features. In a cytochemical analysis, the blasts were positive for myeloperoxidase (MPO) but negative for α-naphthyl butyrate esterase (ANBE) and periodic acid Schiff (PAS) stains. All erythroid precursors were negative for PAS. A few blasts with cytoplasmic vacuoles were negative for MPO and PAS stains. Flow-cytometric immunophenotyping of the blasts showed positivity for CD34 and markers of myeloid lineage, such as CD13, CD15, CD33, CD117, and CD65, with aberrant expression of CD2 and CD19 (Table 1). Immunophenotypically, the blasts were positive for both myeloid and some lymphoid markers, such as CD19, but a population of blasts was not divided into two populations by immunophenotypic analysis. Cytogenetic analysis of the patient's BM cells showed characteristics of 41,XY,-5,-7,add(11)(q23),der(14;21)(q10; q10),-16,add(17)(p13),-18,-20,-21,-22,+2mar[20]. A BM biopsy showed 60% cellularity and several nodules composed of leukemic blasts.

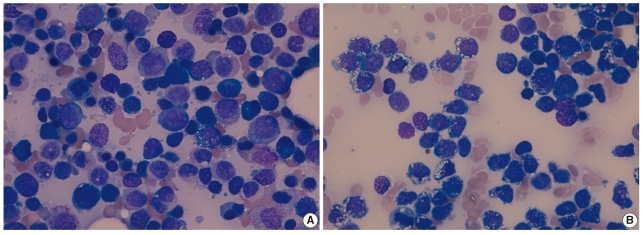 | Fig. 1Findings of the bone marrow aspirate analysis (Wright stain,×1,000). (A) Erythroleukemia (AML-M6a) at initial diagnosis showed medium-sized myeloblasts with a moderate amount of cytoplasm without vacuoles, although a few blasts contained cytoplasmic vacuoles. (B) Precursor B cell ALL at relapse showed lymphoblasts that contained cytoplasmic vacuoles.
|
Table 1
Laboratory findings of the patient at diagnosis and at relapse
*'Positive' was defined as stained blasts that showed higher fluorescence than isotype control. †Intermediate: fluorescence intensity of CD45 for blasts.
Abbreviations: MPO, myeloperoxidase; PAS, periodic acid Schiff; ANBE, α-naphthyl butyrate esterase; TDT, terminal deoxynucleotidyl transferase; IHC, immunohistoche-mistry; FLT3-ITD, fms-like tyrosine kinase 3-internal tandem duplications; TKD, tyrosine kinase domain.
![]()
We made a diagnosis of erythroleukemia and started induction chemotherapy with cytarabine and daunorubicin. After the first course of induction treatment, the patient's BM analysis still showed 9.6% blast cells; therefore, we performed reinduction therapy with cytarabine and daunorubicin. Two months after the initial diagnosis, complete remission was achieved and cytogenetic analysis of the patient's BM showed a normal karyotype of 46,XY[20]. Consolidation chemotherapy was continued with cytarabine, and the remission was maintained for 2 months.
Four months after the initial diagnosis, the patient presented with peripheral leukocytosis, anemia, and thrombocytopenia. Peripheral blood analysis yielded the following results: hemoglobin, 9.4 g/dL, platelet count, 20×109/L, and leukocyte count, 15.5×109/L, with 30% blasts. In the BM analysis, 91.2% of all nucleated cells were blasts. At relapse, the blasts had large, irregularly shaped nuclei with fine chromatin, inconspicuous nucleoli, and a moderate amount of light blue cytoplasm with many vacuoles (Fig. 1B). The small proportion of blasts with cytoplasmic vacuoles at the initial diagnosis became predominant in the relapse. The blasts did not stain with the MPO and ANBE stains, but they showed positive staining with the PAS stain, with a coarse, granular positivity pattern. Immunophenotyping of the blasts revealed positivity for CD34, CD13, CD15, CD19, cytoplasmic CD22, terminal deoxynucleotidyl transferase (TDT), CD65, and CD79α and absence of anti-myeloperoxidase (Table 1). The karyotype of the BM cells in the relapsing condition was 41,XY,add(1)(q21),-3,add(3)(q22),-4,-5,-7,der(9)t(9;11)(q34;q13),der(11)del(11)(p11.2)add(11)(q23), der(14;21)(q10;q10),-16,add(17)(p13),-18,-20,-22,+4mar[20]. A BM biopsy showed 70% cellularity, with most cells being leukemic blasts.
A diagnosis of precursor B ALL was made on the basis of the 2008 WHO classification system. Chemotherapy with vincristine and prednisolone was performed. Fourteen days after relapse, the patient's BM still showed 20% cellularity with persistence of blasts among nucleated cells. Unfortunately, chemotherapy was discontinued because of pneumonia and sepsis, and he died of progressive pneumonia.
Go to :
DISCUSSION
Although conversions from ALL to AML have been reported, lineage switches from AML to ALL are rare (Table 2) [1, 6-14]. Further, to our knowledge, this is the first study to report a case of conversion from erythroleukemia to ALL.
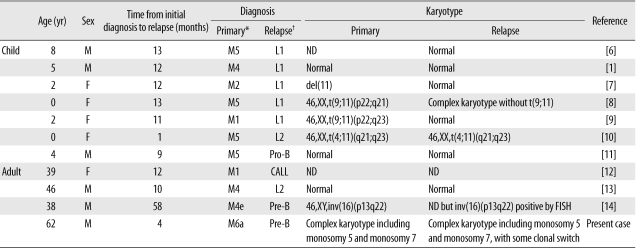
Table 2
Reported cases of lineage switches from AML to ALL
*Primary leukemia was diagnosed according to the FAB classification. †Relapsing leukemia was diagnosed according to the FAB or WHO classification.
Abbreviations: M, male; F, female; ND, not done; Pro-B, pro-B lymphoblastic leukemia; CALL, common cell ALL; Pre-B, precursor B-cell lymphoblastic leukemia.
![]()
Several hypotheses have been proposed to explain lineage switches in acute leukemia [13, 15-17]. According to the common myeloid/lymphoid progenitor cell hypothesis, uncommitted progenitor cells that have both early myeloid and T- or B-lymphoid markers lose 1 lineage marker during the maturation process and differentiate toward the other lineage [15]. This hypothesis has been supported by the spontaneous lineage switches in the blast transformation phase of chronic myeloid leukemia [16]. According to the primary chemotherapy-related hypotheses, the predominant leukemic clone may be suppressed by chemotherapy, allowing the expansion of subclones with different phenotypes [13, 17].
According to the secondary chemotherapy-related hypotheses, AML may arise because of the cytotoxic effects of conditioning regimens and chromosomal abnormalities, particularly of chromosomes 5 and 7, and 11q23 translocations that occur preferentially during the malignant transformation of pluripotent stem cells [18]. In some cases, topoisomerase II inhibitors (epipodophyllotoxins and anthracyclines) affect secondary AML; for example, secondary AML occurring after anthracycline treatment of a patient with acute promyelocytic leukemia has been reported to be associated with cytogenetic abnormalities involving chromosomes 5 and 7 [18]. Secondary ALL, however, is very rare and is associated with only a few fusion genes, such as IgA and T-cell receptor gene rearrangements [18].
Our patient was treated with cytarabine and daunorubicin (anthracycline) and had chromosomal abnormalities involving chromosomes 5 and 7 during both the initial diagnosis and the relapse. A few additional cytogenetic abnormalities, such as monosomy 3 and 4, appeared after the lineage switch, indicating that a treatment-related lineage switch had occurred. However, the leukemic blasts at initial diagnosis already showed 2 kinds of morphology, and the small proportion of blasts observed at the initial diagnosis grew, becoming predominant at relapse. Moreover, the patient's leukemia relapsed with lineage conversion only 4 months after the initial diagnosis, with the same leukemic clones showing complex karyotypic abnormalities. These findings suggest that a previously existing small population of lymphoid progenitor cells expanded during relapse. However, the small portion of blasts observed at the primary diagnosis was negative for PAS stain, and the immunophenotype of the blasts was not representative of a mixed populations of blasts. Thus, these findings were more supportive of the dedifferentiation of common myeloid/lymphoid progenitor cells.
In most patients with a lineage switch, leukemic blasts observed at relapse differ in morphology from the primary blasts; this may include differences in cell size, amount of cytoplasm, or presence of Auer rods. In rare cases of lineage switch, however, the primary and relapsed blasts have a similar morphology [19]. Most patients who had leukemia with lineage switch showed relapse after 1-4 yr from the initial diagnosis of leukemia [1]. In some cases, very early relapse with lineage switch had been reported only 3 months after the initial diagnosis [3].
It is not always possible to perform the full panel of immunophenotyping assays in cases of relapsing leukemia; therefore, the frequency of the lineage switch phenomena could be underestimated. The treatment of AML is completely different from that of ALL. Thus, it is important that sufficient immunophenotypic markers for relapsing leukemia be employed to arrive at the correct diagnosis. Correct diagnosis and early recognition of lineage switch is important for effective therapeutic intervention.
Go to :
 XML Download
XML Download