

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Miller-Dieker syndrome (MDS) is a multiple-malformation syndrome in which the patients show severe lissencephaly with facial dysmorphism at birth. Lissencephaly varies in severity from agyria to pachygyria and is linked to defects in the lissencephaly gene (LIS1), which is located in 17p13.3 [1]. In patients with MDS, the loss of the LIS1 gene can occur by deletion [2], ring chromosome formation [3], or transmission of a parental chromosome with an unbalanced translocation, including transmission of chromosome 17p13.3 [4]. In approximately 80% of the cases of MDS in infants, the disease is believed to be caused by a de novo deletion, and the remaining cases are caused by inheritance [5]. In the case of a parental balanced translocation, the chromosomes of the offspring usually show an unbalanced translocation combined with partial trisomy of another autosomal segment.
Various 17p13.3 microdeletions inherited from a parent with a balanced translocation of chromosome 17 and another chromosome have been reported [4, 6]. Most patients with an unbalanced translocation show a combination of the findings of MDS and additional unique phenotypes because of the involvement of segments of various autosomes.
In this report, we report the findings for a female infant who had der(17)t(12;17)(q24.33;p13.3)pat showing a nearly pure large deletion (~4.6 Mb) involving 17p13→pter without evidence of duplication of 12q24.33→qter, which was confirmed by high density array comparative genome hybridization (CGH). Recently, Grosso et al. [7] also reported a case of MDS with periventricular nodular heterotopia caused by an unbalanced t(12;17)(q24.31;p13.3) translocation. Their case had a relatively large duplication (~11 Mb) of 12q24.31→qter, but our case showed no definite duplication of 12q in the array CGH analysis.
Our case could have been misdiagnosed as a classic case of MDS resulting from the deletion of 17p13.3→pter because the smallest segment of 12q24.33→qter according to the literatures was involved. If such patients are incorrectly diagnosed with MDS caused by de novo 17p13.3 deletion, the resultant improper genetic counseling may make it difficult to exactly predict the potential risk of recurrent lissencephaly for successive pregnancies.
Go to : 
CASE REPORT
1. Clinical presentation
The female patient was born to a healthy G4P2 mother and a healthy non-consanguineous father at 35 + 5 weeks of gestation by cesarean delivery because of preterm labor and intrauterine growth retardation. The parents' first child was diagnosed with lissencephaly after birth and died at 3 months of age. The second baby was aborted following the diagnosis of a brain anomaly by fetal ultrasound.
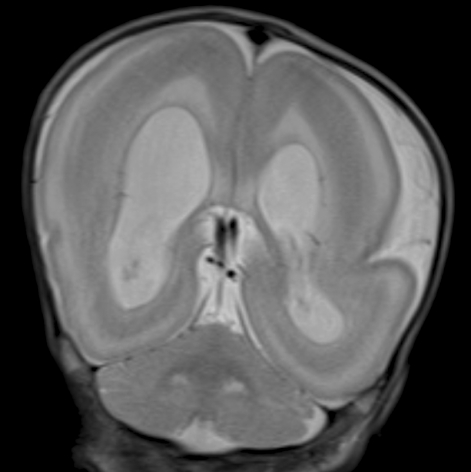
The patient was their third baby and was diagnosed with ventriculomegaly during the antepartum period. The birth weight was 1,680 g (<3rd percentile), the length of the baby was 42.3 cm (<3rd percentile), and the head circumference was 30 cm (3rd-10th percentile). At birth, she was noted to have facial dysmorphism, including prominent frontal bossing, a high nasal bridge, low-set ears, a thin inferior lip, micrognathia, and arthrogryposis. On the 3rd day after birth, she was diagnosed with symptomatic patent ductus arteriosus by echocardiography. Neuromotor delay with marked generalized hypotonia was observed. An electroencephalogram showed diffuse brain dysfunction, and magnetic resonance imaging (MRI) showed lissencephaly with band heterotopia and agenesis of the corpus callosum (Fig. 1). At 5 months of age, she was taken to the emergency room because of generalized tonic seizures and showed failure to thrive. The parents had no history of epilepsy or neurologic problems.
2. Cytogenetic and molecular studies
Conventional cytogenetic analysis of the proband revealed a possible abnormal karyotype [46,XX,del(17)(p13)] (Fig. 2A). Cytogenetic analysis was also performed on the proband's parents because the recurrent abnormal phenotypes during the 3 consecutive pregnancies strongly suggested that MDS in this case might have resulted from an unbalanced segregation of the parental balanced translocation of 17p with other chromosomes. The mother's karyotype was normal.
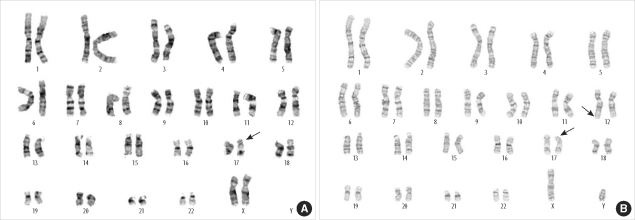
Cytogenetic analysis of the proband's father showed a shortened p-arm of chromosome 17 (Fig. 2B), which strongly suggested a balanced translocation of 17p and another chromosome. However, the small translocated telomeric segments of chromosome 17 could not be definitively identified by conventional cytogenetic analysis.
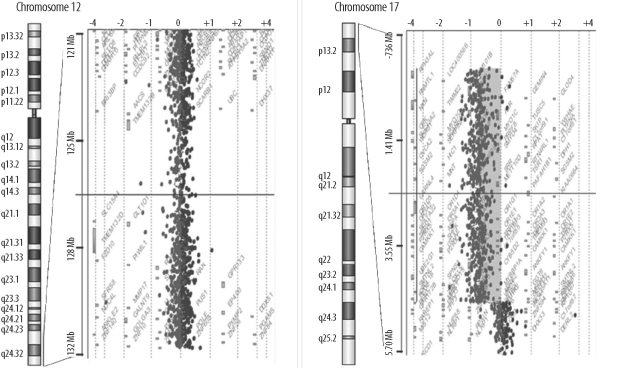
In order to identify the position where the terminal segment of chromosome 17p13.3→pter was translocated, FISH with a Miller-Dieker/lissencephaly region probe (Vysis, Abbott Laboratories, Abbott Park, IL, USA) was performed on the same G-banded metaphase chromosomes according to the manufacturer's instructions. The MDS probe signal (17p13.3) was detected on chromosome 12qter, indicating an unbalanced segregation of t(12;17) (Fig. 3). Further characterization of the size and boundaries of the cytogenetic abnormalities of the proband was performed using Human Genome CGH Microarray Kit 244 (Agilent Technologies, Palo Alto, CA, USA), which consists of approximately 244,000 arrayed 60-base oligonucleotide-probes that span both coding and non-coding sequences with an average spatial resolution of about 15 kb. Array CGH showed about a 4.6-Mb deletion involving 17p13.3, but it did not show duplication at 12q [arr17p13.3p13.2 (28,969-4,653,156)×1 pat] (Fig. 4).
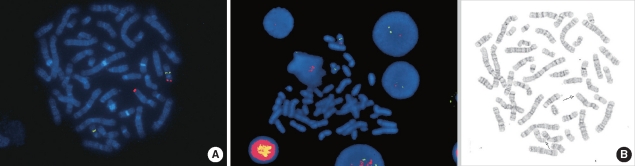 | Fig. 3(A) Proband's metaphase FISH results. One orange and 2 green signals indicate loss of 17p13.3. (B) Sequential G-banding (left) and FISH (right) performed on her father's metaphase chromosomes. G-banded metaphase with arrows indicating chromosome 12 and 17. Note that an orange signal of 17p13.3 is observed in chromosome 12q.
|
Go to :
DISCUSSION
Our case showed a deletion involving 17p13.3→pter and a severe clinical phenotype of MDS, including lissencephaly with band heterotopia, corpus callosum agenesis, peculiar facial morphology, cardiac defects, and limb anomalies. The deletion in 17p13.3→pter in our case included simultaneous deletion of the platelet-activating factor acetylhydrolase 1b, regulatory subunit 1 (PAFAH1B1) gene (previously called LIS1), and the tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, epsilon polypeptide (YWHAE). Lissencephaly is associated with the loss or mutation of the PAFAH1B1 gene, which contributes to neuronal migration during embryological development, and additional loss of the v-crk sarcoma virus CT10 oncogene homolog (avian) (CRK) and YWHAE genes. These genes are telomeric to the PAFAH1B1 gene and contribute to cortical development and the severity of MDS [8].
Grosso et al. [7] reported a case of MDS with periventricular nodular heterotopia caused by an unbalanced t(12;17) (q24.31;p13.3) translocation, which showed a 3.7-Mb deletion at 17p13.3→pter and a duplication of about 11 Mb at 12q24.31→qter. Our case showed a 4.6-Mb deletion involving 17p.13.3→pter and no identifiable duplication at 12q24.33. The phenotypes of our case were similar to those of the case reported by Grosso et al. [7], except for cortical heterotopia. Cortical heterotopias are relatively uncommon in MDS, but if present, they are usually in the form of subcortical heterotopias [9]. Periventricular heterotopias have been described in MDS. However, the YWHAE gene has been recently reported to contribute to periventricular heterotopic malformation [10].
Our case showed no definite duplication of 12q in the high-resolution array CGH, and there were no recognizable phenotypic features of a partial duplication of 12q24.33→qter. A phenotype for trisomy 12q has been described on the basis of the size differences of the duplicated 12q. Chromosome 12q duplication syndrome has common clinical features, including mild development delay, mild mental retardation, broad forehead, simplified ear helices, small mouth, and thin lips [11]. Partial trisomy of 12q24.33→qter with a 1.3-Mb chromosome imbalance derived from a familial t(12;17)(q24.33;q25.3) showed only some mild phenotypic similarities with the features of the 12q duplication syndrome [12]. Sathanoori et al. [13] reported the smallest cryptic duplication (~423 kb) of 12q24.33→qter, in which about 37 genes were found to be listed in the human genome database (Ensembl v37). The genes included GOLGA3, CHFR, and zinc finger genes (ZNF605, ZNF26, ZNF84, ZNF140, ZNF10, and ZNF268). However, no important genes in the 12q24.33→qter region were identified in our case.
In comparison with bacterial artificial chromosome (BAC) arrays, the high-resolution oligonucleotide arrays used in our case offer a much greater probe density for better detection of smaller genomic imbalances with a functional lower limit of 36 kb [14]. Thus, chromosomal alternations smaller than ~36 kb may be theoretically undetectable by array CGH (Agilent Technologies). In our case, even if the cryptic region of 12q is not detected by high-resolution array CGH, the chromosomal alterations would be considered benign or clinically insignificant because of the absence of recognizable phenotypic features of a partial duplication of 12q24.33→qter in the karyotype. Moreover, a consensus statement for array CGH recently described that the smallest known recurrent pathogenic microdeletion syndromes are ~500 kb or larger [15], and array CGH, with a lower limit of resolution of ~400 kb throughout the genome, will reliably identify all known recurrent pathogenic microdeletion and microduplication syndromes [15, 16]. In addition, although cytogenetic events might appear to be balanced at the microscopic level of resolution, many such events have submicroscopic imbalances [17].
In our case, the MDS caused by a balanced translocation of t(12;17) was difficult to detect and would have gone unnoticed without the previous history of recurrent lissencephaly. The use of FISH on giemsa-trypsin-leishman (GTL) banded metaphase chromosomes (sequential G-banding and FISH) is effective for defining the cryptic translocation. Moreover, array CGH has enabled further delineation of the clinical spectrum of known chromosomal abnormalities and the identification of novel, recurrent genetic imbalances [18].
In summary, this is a rare MDS case derived from a parental balanced translocation confirmed by genomic array CGH and FISH. For this reason, the deletion of 17p in MDS in the proband should be not interpreted as a de novo deletion without a parental study, especially in parents with a history of recurrent pregnancy loss. Accurate genetic counseling can be offered to the family in cases where there is a potential risk of recurrent lissencephaly in the next pregnancy. The combined use of array CGH and FISH analysis is a powerful tool for the diagnosis of genetic diseases and can reveal unexpected genomic imbalances.
Go to :
 XML Download
XML Download