

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Molecular typing methods have been extensively applied to understand the genetic variations in and phylogenetic relationships among various Mycobacterium tuberculosis strains, particularly the strains responsible for large-scale infection outbreaks and the drug-resistant strains, and to differentiate re-infection from relapse [1-3].
Detection of the IS6110-restriction fragment length polymorphism (RFLP) is considered the gold standard for genotyping M. tuberculosis, because this RFLP has high discriminatory power [4]. However, RFLP has some drawbacks; a long culture time is required to obtain enough DNA, and it is time- and labor-intensive [5]. Unlike the IS6110 genotyping method, spoligotyping, which is based on polymorphisms of direct repeat loci, is simple and rapid because it is PCR-based [6, 7]. One limitation of this method is its inability to discriminate Beijing family strains. This renders the method unsuitable for strain typing as an epidemiologic tool in Korea where the Beijing family strains are predominant [8, 9].
Recently, genome sequence data of 4 M. tuberculosis complex strains have become available, and this has enabled the development of a molecular method using single-nucleotide polymorphisms (SNPs) to identify M. tuberculosis strains [10, 11]. These SNPs can be used as a precise tool in phylogenetic studies [12-14]. SNP analysis also provides a powerful strategy for large-scale molecular population studies examining phylogenetic relationships among bacterial strains [15]. However, there is no standard SNP analysis method for estimating genetic relationships between strains isolated during large-scale outbreaks of infection or drug-resistant strains.
Mycobacterial interspersed repetitive units-variable number of tandem repeats (MIRU-VNTR), another strain-typing method, has been developed in the past decade [2]. This method is considerably faster, requires only small amounts of DNA, and can be easily digitized to share data among laboratories [5, 16]. Thus, SNP and MIRU-VNTR analyses are good alternative methods for M. tuberculosis molecular strain typing.
This study applied SNP and MIRU-VNTR analyses to M. tuberculosis isolates collected throughout Korea.
Go to : 
MATERIALS AND METHODS
1. Bacterial isolates
We studied 102 clinical M. tuberculosis isolates collected from 11 university hospitals in Korea in 2008 and 2009. Among the isolated strains, 96 were collected from single cultures performed in 96 patients, and 6 were collected from different cultures performed in the same patients. Epidemiologic distribution of the 96 isolates had already been characterized by IS6110-RFLP, and all isolates were collected from epidemiologically unrelated patients [17]. The 6 paired strains were used to evaluate the reproducibility of the strain-typing methods used in this study.
2. SNP analysis
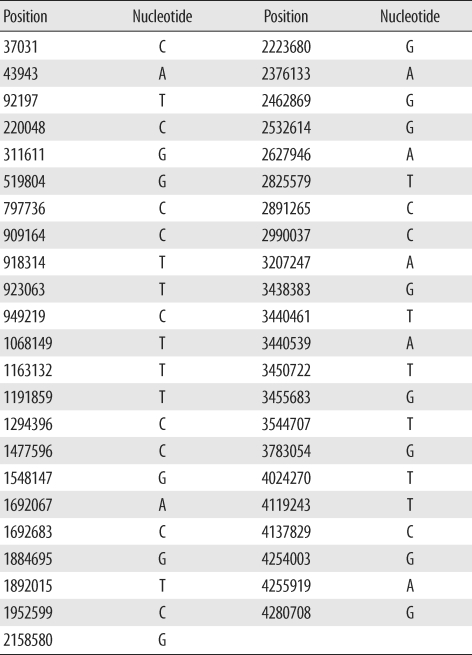
We selected 45 SNPs previously demonstrated to provide high resolution between clinical isolates [18]. Positions of each of the 45 SNPs and its nucleotides in M. tuberculosis H37Rv are presented in Table 1. SNPs were detected using hairpin primer (HP) assays as described previously [19]. DNA including a target nucleotide in each primer was amplified using the HP assay. A wild-type HP primer and different mutant HP primer were used in a complementary fashion. Primer sequences are published in a previous paper and are not repeated here [19]. In brief, the amplification protocol was as follows: stage 1, 95℃ for 10 min, 70℃ for 30 sec; stage 2, 72℃ for 30 sec, 95℃ for 20 sec, 69℃ for 30 sec, lowering 1° in the last step for every cycle during 10 cycles; and stage 3, 72℃ for 30 sec, 95℃ for 20 sec, and 60℃ for 30 sec; this was repeated 40 times. PCR products were analyzed on a 2% agarose gel.
3. MIRU-VNTR
MIRU-VNTR was performed as described previously [20, 21]. Briefly, isolates were genotyped by PCR amplification of the 12 MIRU-VNTR loci (MIRU-02, -04, -10, -16, -20,-23, -24, -26, -27, -31, -39, and -40) and 4 exact tandem repeat (ETR) loci (ETR-A, -B, -C, -F). The amplification protocol consisted of 30 cycles of 30 sec at 95℃, 30 sec at 61℃, and 1 min at 72℃. PCR products were analyzed on a 2% agarose gel, and the number of tandem repeats was calculated.
5. Phylogenetic analysis
Phylogenetic analysis was performed using the complete data of 96 isolates for SNP analysis at 45 loci and for MIRU-VNTR analysis at 16 loci. A dendrogram of SNP and MIRU-VNTR analyses was constructed from the UPGMA cluster based on the overall similarity of each nucleotide polymorphism (http://genomes.urv.cat/UPGMA/). For discrimination analysis of MIRU-VNTR, the Hunter-Gaston diversity index (HGDI) was calculated as described elsewhere [22] and used for comparison of the discriminatory power of each VNTR locus.
Go to :
RESULTS
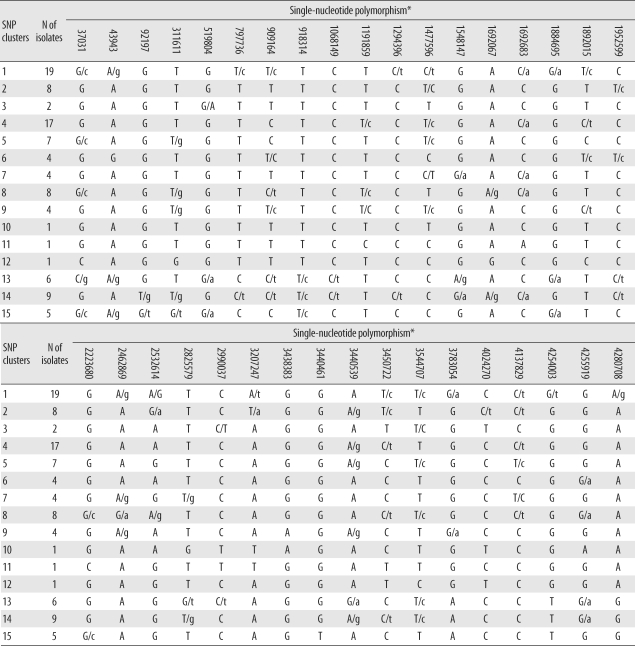
1. Evaluation and phylogenetic analysis of SNPs
Among the 45 SNPs, 35 had polymorphisms that could be used to differentiate the 96 tested isolates. The other 10 SNPs did not contain polymorphisms. Based on SNP results, isolates were classified into 15 clusters (Fig. 1). Cluster 1, the most populated cluster, contained 19 isolates, followed by cluster 4 with 17 isolates. Three clusters consisted of only 1 isolate each (Table 2). As shown in Fig. 1, nearly all Beijing family strains, which are defined and identified by IS6110 RFLP data [17], were distributed within closely related clusters in the SNP dendrogram. Seventy-four of the 76 Beijing family strains were allocated to SNP clusters 1 through 12. The remaining 2 isolates were allocated to clusters 13 and 14. Nearly all 14 isolates of the K family, a subfamily of the Beijing family identified in a previous study [17], were allocated to clusters 4 and 6.
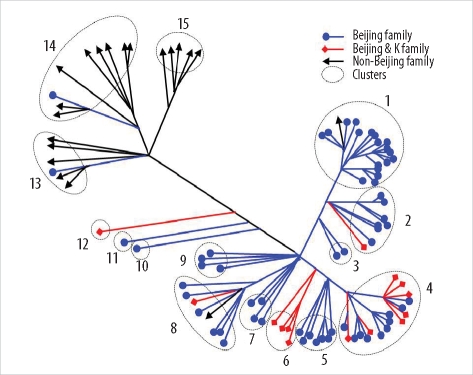 | Fig. 1Distance-based neighbor joining phylogenetic tree of 96 Mycobacterium tuberculosis isolates based on 35 single-nucleotide polymorphisms reveals 15 clusters (indicated by dotted circles). Each IS6110-restriction fragment length polymorphism type is indicated by a closed circle, quadrangle, or arrow.
|
2. Phylogenetic analysis of VNTR
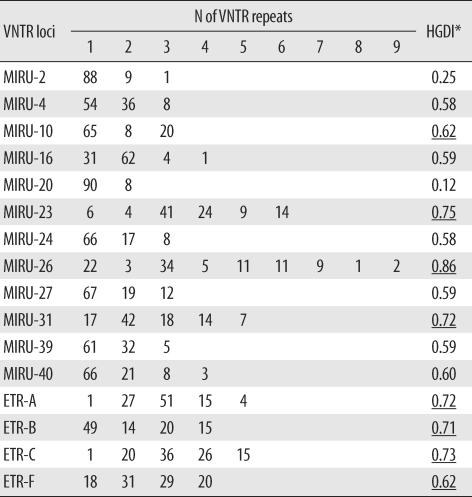
The 96 isolates showed amplification products from 16 VNTR loci and contained at least 1 copy of each locus. Results of allelic diversity and HGDI testing are summarized in Table 3. Among the 16 loci, the discriminatory index in 8 (MIRU-10, -23, -26 and -31; ETR-A, -B, -C, and -F) was high (HGDI > 0.6) according to the definition of Sola et al. [23]. Six loci (MIRU-4, -16, -24, -27, -39 and -40) were dispersed and discriminated moderately well (0.3 ≤ HGDI ≤ 0.6). Two loci (MIRU-2 and -20) were poorly discriminated (HGDI < 0.3). The complete data were divided into 12 groups. The largest group was group 2, which included 15 isolates. Group 11 contained the only unique pattern (Fig. 2). Unlike the SNP method, MIRU-VNTR did not show notable localizations of Beijing or non-Beijing family isolates in specific clusters.
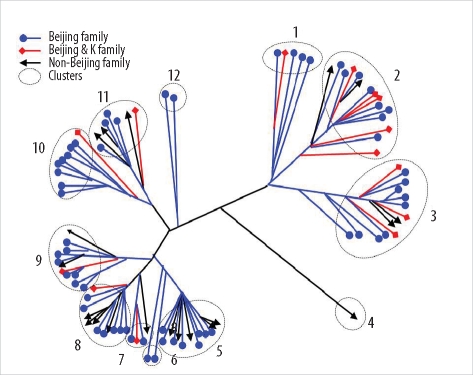 | Fig. 2Distance-based neighbor joining phylogenetic tree of 96 Mycobacterium tuberculosis isolates based on based on mycobacterial interspersed repetitive units-variable number of tandem repeats of 16 loci reveals 12 clusters (dotted circles). Each IS6110-restriction fragment length polymorphism type is indicated by a closed circle, quadrangle, or arrow.
|
Table 3
Variable repeat numbers in each VNTR locus and allelic diversity among 96 clinical isolates of Mycobacterium tuberculosis collected throughout Korea
*Underlining indicates loci showing high discriminatory power, as defined by Sola et al. [23].
Abbreviations: VNTR, variable number of tandem repeats; HGDI, Hunter-Gaston diversity index; MIRU, mycobacterial interspersed repetitive units; ETR, exact tandem repeat.
![]()
3. Reproducibility of SNP and VNTR analyses
The 6 paired isolates produced exactly the same results in IS6110-RFLP, SNP, and VNTR patterns for a given patient (data not shown), demonstrating that the 2 methods are reproducible.
Go to :
DISCUSSION
SNP analysis was developed after whole-genome sequence data became available and has been for genome-scale analysis to estimate overall chromosomal relationships among isolates [24, 25]. SNP analysis is a powerful strategy for examining phylogenetic relationships among bacterial strains and is easy to perform [26, 27]. Among various methodologies described previously, we chose the HP assay for this study to improve PCR specificity by decreasing nucleotide mismatching [19, 28]. Additionally, the material costs are lower for this method than for a sequencing-based method [19]. Previous studies have demonstrated that all tested M. tuberculosis strains were distinguishable by SNP analysis, and therefore, the authors proposed that SNP analysis was an ideal tool to investigate biological variations among clinical M. tuberculosis isolates [15, 18]. Filliol et al. [18] identified a minimal number of 45 highly informative SNPs by analyzing 212 SNPs against 323 clinical M. tuberculosis and Mycobacterium bovis isolates. Our SNP analysis was based on this set of 45 SNPs. In this study, however, only 35 SNP loci could be used to categorize our 96 M. tuberculosis isolates. Interestingly, nearly all Beijing family strains, including those of the K family, were allocated to closely related clusters. Our results indicate that SNP analysis using 35 loci is useful for phylogenetic analysis of clinical M. tuberculosis isolates from Korea. This method is effective, easy, and economical. In the 96 M. tuberculosis isolates examined, 10 of 45 SNPs were identical. We think that this difference was due to the difference in strain distribution between the current and previous studies [18]. Our study included a larger proportion of Beijing family isolates, whereas only 15 Beijing family isolates, among the 246 tested isolates, were included in the previous study [18]; moreover, those 15 isolates were classified as a single SNP group. Therefore, some SNPs proven to be polymorphic between strains isolated in 1 region may not be suitable to discriminate strains originating in other regions.
The MIRU-VNTR typing method is technically more flexible and faster than IS6110-RFLP typing, and the results are expressed as numerical codes, which are easy to compare and exchange [29, 30]. A previous report demonstrated that MIRU-VNTR analysis using well-selected loci is useful for analyzing clinical M. tuberculosis isolates from Korea, where the Beijing family is predominant [31]. In the current MIRU-VNTR study using 96 isolates, we identified 2 interesting findings. First, in contrast to SNP results, MIRU-VNTR patterns were not related to IS6110-RFLP patterns. As shown in Fig. 2, isolates of Beijing and non-Beijing families were distributed in almost all clusters. This means that changes in MIRU-VNTR loci developed independently of changes in IS6110 insertion. Hanekom et al. [32] described discordant results between MIRU-VNTR and IS6110 RFLP and suggested that the degree of discordance was dependent on the genetic distance between isolates. However, SNP analysis resulted in clusters closely related to IS6110-RFLP patterns. Filliol et al. [18] described similar close clustering of Beijing family strains by both the SNP and spoligotyping methods. We could not locate M. tuberculosis molecular strain-typing data that had been obtained by simultaneously using IS6110-RFLP, SNPs, and MIRU-VNTR. Here, we describe a close relationship between SNP and IS6110-RFLP and independent results for MIRU-VNTR and IS6110-RFLP. This means that, unlike IS6110-RFLP, spoligotyping, or SNP, MIRU-VNTR shows high discriminatory power for typing M. tuberculosis strains in areas where the Beijing family is highly prevalent. Second, the discriminatory power of MIRU-VNTR in some loci varied from that described in a previous study conducted with 81 isolates from Korea [31]. In that study, Yun et al. [31] reported that MIRU-23 and ETR-A, -B, and -C had low discriminatory power (HGDI < 0.3). In our study, however, these 4 loci were highly discriminatory (HGDI > 0.6), with MIRU-23 having the second highest discriminatory power (HGDI = 0.75). The highest discriminatory power was shown by MIRU-26 in both studies (HGDI > 0.80). The discriminatory power of a specific locus is inevitably affected by the selection of isolates, and the selection criteria were different between the 2 studies. In the previous study, all isolates were collected from Pusan National University Hospital or provided by the Korean Institute of Tuberculosis, so the distribution of patients' place of residences might have been restricted [31]. In this study, all 96 isolates were collected from 11 university hospitals located in various areas, and the number of isolates from each hospital was roughly proportional to the recorded patient numbers in the area served by that institution. Therefore, our study represents MIRU-VNTR performance more accurately. Combining the results of both the studies, we found that MIRU-10, -26, -31, and ETR-F show a high discriminatory power for molecular strain typing of M. tuberculosis isolates from Korea. Our assessment of the clinical utility of other loci, such as ETR-A, -B, and -C, that showed good discriminatory power in only 1 study is reserved until further data are obtained.
We confirmed the reproducibility of SNP and MIRU-VNTR analyses using 6 pairs of strains from different cultures of the same patients. Both SNP and MIRU-VNTR can discriminate clinical M. tuberculosis isolates in Korea, where Beijing family strains are highly prevalent. Both methods are PCR-based and early application to clinical specimens is possible. Strain typing is often requested to detect outbreaks or contamination, and rapid results are required to control these phenomena. Although IS6110-RFLP has high discriminatory power, it is not useful for these purposes. Therefore, SNP and MIRU-VNTR can be used as surrogate molecular strain-typing methods for M. tuberculosis in Korea.
Go to :
 XML Download
XML Download