

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Inherited disorders of Hb are classified into thalassemias and structural variants [1]. Thalassemia is characterized by defects in the synthesis of a globin chain, and the majority of thalassemias involve α- or β-globin chains [2, 3]. Structural variants, called hemoglobinopathies, are structural abnormalities of Hbs [4].
The β-thalassemia trait is associated with mild or no anemia but with reduced mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH) values, and an elevated HbA2 level [5]. β-thalassemia is uncommon in the Korean population; however, it must be considered in the differential diagnosis of hypochromic anemia [6].
HbA2 has been measured using cellulose acetate (alkaline) or citrate agar (acid) electrophoresis, isoelectrofocusing (IEF), microcolumn chromatography, and high-performance liquid chromatography (HPLC) [7]. Electrophoresis is the main tool used for the identification and quantification of variant Hbs. This provides a clear background, but it is not possible to differentiate between HbE and HbO, and between HbD and HbG [8]. Moreover, electrophoresis is time-consuming, labor-intensive, and inaccurate in the quantification of low-concentration Hb variants or in the detection of fast Hb variants [8]. Although IEF has excellent resolution, it has the same disadvantages as other electrophoretic methods [7, 8]. Column chromatography is satisfactory for β carrier diagnosis; however, it is also laborious, intensive, and time-consuming [7]. In addition, HbA2 increases slightly in the presence of unstable Hb [7].
HPLC is the method of choice for the initial screening of Hb variants and for quantification of Hb fractions. However, its use has not become widespread in clinical laboratories since it requires special instrumentation and training, and the results appear in complex patterns [9]. Consequently, many clinical laboratories still use alkaline and acid gel electrophoresis to screen for hemoglobinopathies. Additionally, HPLC has some limitations, including falsely decreased HbA2 levels in patients with the HbD Punjab trait, falsely increased HbA2 levels in patients with HbS, and co-elution of various Hbs, including HbE, Hb Osu Christianborg, HbG Coushatta, and Hb Lepore with HbA2 [10].
Capillary electrophoresis (CE) has been presented as an alternative tool capable of separating the normal Hbs (A, F, and A2), and detecting the major Hb variants by alkaline electrophoresis on silica capillaries [11]. It can achieve simultaneous analysis, fast separation, good resolution, high accuracy, and full automation. Furthermore, this method is better able to separate HbA2 from HbE, HbC, Hb Lepore, and HbS than the HPLC method [7]. As the use of CE expands for evaluation of thalassemia and hemoglobinopathies, it may help reveal the characteristics and prevalence of thalassemia mutations in the Korean population that is different from those of high prevalence area.
In this study, Hb fractions were measured in patients with hypochromic microcytosis to detect thalassemia and Hb variants and CE was compared with cellulose acetate electrophoresis (CA) to replace CA by CE in a clinical laboratory. To the best of our knowledge, this is the first comparative study of CE and CA for the screening of hemoglobinopathies in Korea.
Go to : 
MATERIALS AND METHODS
1. Samples
Thalassemias and hemoglobinopathies were evaluated in 143 adult blood samples collected in EDTA tubes. Among these, 51 came from male donors and 92 from female donors. The median age was 47 yr (range, 20-89 yr). Forty samples were obtained from normal individuals during a regular health check-up. One hundred and three samples were obtained from patients with hypochromic microcytosis. The microcytic hypochromic group consisted of patients with an MCV of less than 75 fL and an MCH less than 24 pg with or without anemia [7]. Patients having conditions such as acute and chronic inflammatory diseases, infections, thyroiditis, acute bleeding, or any type of malignancy that might affect ferritin or Hb levels were excluded. Complete blood cell counts (CBC), CE, and CA were performed on each sample.
2. Hematologic studies
The CBCs were determined by an automated blood cell analyzer XE2100 (Sysmex, Kobe, Japan) within 3 hr after blood collection. Serum iron levels, total iron-binding capacities (TIBC), and serum ferritin levels were measured in each case. Serum iron levels and TIBC were measured using a Toshiba 200FR autoanalyzer (Toshiba Co., Tokyo, Japan). Serum ferritin levels were evaluated by an Architect system (Abbott Laboratories, Abbott Park, IL, USA).
3. Cellulose acetate electrophoresis
The Helena electrophoresis system (Helena Laboratories, Beaumont, TX, USA) was used to electrophorese hemolysates on Titan III cellulose acetate plates at 350 V for 25 min in an alkaline buffer. The results were analyzed according to manufacturer's guidelines.
4. Capillary electrophoresis
The CE was performed using the Minicap system (Sebia, Norcross, France) according to the manufacturer's instructions, running controls with every test as described previously [9]. The Minicap system uses the principle of CE in free solution. Charged molecules are separated by their electrophoretic mobility in an alkaline buffer with a specific pH. Separation also occurs according to the electrolyte pH and electro-osmotic flow. This novel system has capillaries in parallel allowing multiple and simultaneous analysis. Electropherograms were expressed with divided zones from Z1 to Z15 based on standardizing the location of HbA.
5. Reference values of the two methods
Manufacturer's reference values were used and the reference values were validated by analysis of samples from 40 outpatients and inpatients exhibiting RBC, Hb, MCV, MCH, and MCHC (mean corpuscular hemoglobin concentration) within the reference ranges.
6. Confirmation
To confirm β-thalassemia, molecular analysis of the β-globin gene was performed in one case. Sequencing was carried out in a reference laboratory.
7. Statistical analysis
Statistical calculations were performed with MedCalc 11.3.0.0 (Med Calc Software Company, Mariake, Belgium). Student's t-test was used to compare RBC parameters and Hb fractions between the control and microcytic hypochromic groups. Comparison of the methods was made with Student's t-test and linear regression. Values of P<0.05 were considered statistically significant.
Go to :
RESULTS
1. Separation of normal and variant hemoglobins by CE
Using CE, we ran the AFSC control (containing HbA, HbF, HbS, HbA2, and HbC) (Fig. 1A). Major hemoglobins, including A, F, S, and C and normal Hbs separated well. In the Minicap system, the pattern of fractions comes out from right to left in the following order: from HbA2 to HbA (Fig. 1B). Glycated fractions were not separated from HbA on CE.
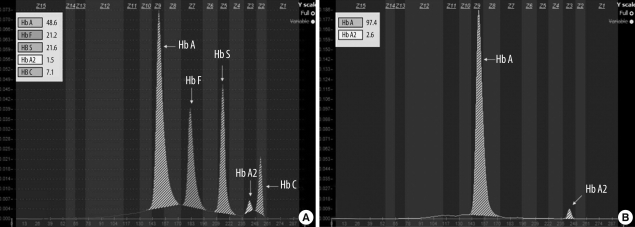
2. Reference values
For CE, the expected values provided by the manufacturer are 96.8-97.8% for HbA, <0.5% for HbF, and 2.2-3.2% for HbA2. Thus, we validated the manufacturer's recommendations and set our laboratory's reference range to 96.8-97.8% for HbA, <1% for HbF, and 2.2-3.5% for HbA2.
For CA, we adjusted the normal ranges for adults to 96.8-97.8% for HbA, <1% for HbF, and 1.9-3.5% for HbA2.
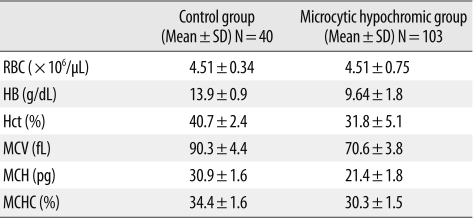
3. CBC and Hb fractions in normal control and microcytic hypochromic group
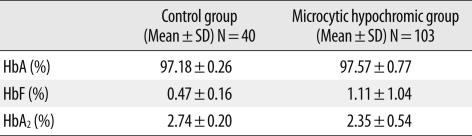
In the microcytic hypochromic group, Hb, Hct, MCV, MCH, and MCHC showed significant differences compared with those of the control group (P<0.0001) (Table 1). There was no significant difference for Hb fraction quantification between the two groups (P>0.05) (Table 2).
4. Comparison of the two methods
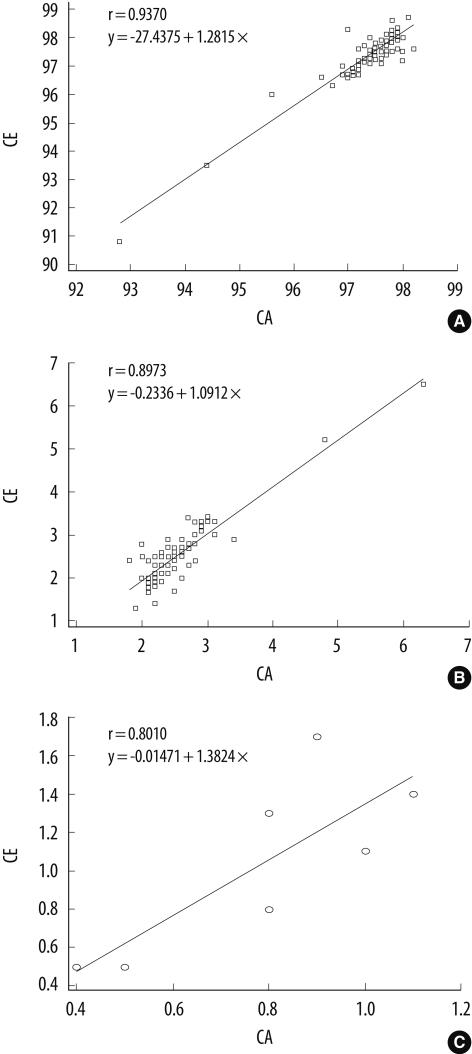
There was a good correlation for measurements of HbA (r=0.9370, P<0.0001), HbA2 (r=0.8973, P<0.0001), and HbF (r=0.8010, P=0.0304) between the two methods (Fig. 2). Both techniques showed similar levels for HbA (97.4±0.7% by CE; 97.4±1.1% by CA) and HbA2 (2.5±0.6% by CE; 2.5±0.7% by CA). The methods also showed comparable results with respect to HbF levels (0.4±1.1% by CE; 0.5±2.7% by CA). No statistically significant difference was found for Hb quantification (P>0.05) (Table 3).
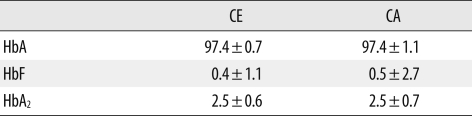
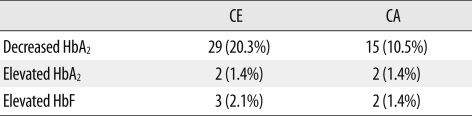
Among the 143 specimens, 29 (20.3%) vs. 15 (10.5%) were found to have decreased HbA2, and 3 (2.1%) vs. 2 (1.4%) had increased HbF for CE vs. CA, respectively. For elevated HbA2, both methods achieved complete agreement in 2 cases (100%). The overall concordance of the 2 methods for Hb fractions was 89.5% (128/143) (Table 4).
5. Cases showing abnormal Hb fractions
The total incidence of abnormal Hb fractions was 1.4% as detected by both CE and CA. In the microcytic hypochromic group, there were 29 cases with decreased HbA2, 2 cases with increased HbA2, 3 cases with increased HbF, and 2 cases with both increased HbA2 and HbF. With the exclusion of the iron deficiency anemia (IDA) patients, 2 patients (1.4%) were suspected to have thalassemia minor. One of these, with an HbA2 level of 6.3%, was confirmed by sequencing. One of the cases with increased HbF had IDA.
Go to :
DISCUSSION
β-thalassemia is primarily found in individuals of Mediterranean, African, and Southeast Asian ancestry [1]. In Korea, approximately 20 cases with β-thalassemia have been reported since the first case report in 1988 [12, 13]. However, with rapid growth in the Southeast Asian segment of the Korean population, the geographic distribution of hemoglobinopathies is expected to become significantly different from what it is today. In this study, CE and CA were used to measure Hb fractions from patients with hypochromic microcytosis to compare their ability to detect thalassemia and Hb variants.
Diagnosis of β-thalassemia can be made by analysis of erythrocyte indices and morphology, and quantification of HbA2 using conventional methods, including HPLC, electrophoresis, and microcolumn techniques [14]. Quantification of HbA2 is challenging because its level is low and increases only slightly in disease, and other Hb variants often interfere with its measurement [15]. CA is routinely used but is inaccurate. HPLC is the method of choice but is costly and not routinely available. Previous studies have shown that the recently introduced CE separates HbA2 well from HbE, HbC, and HbS, with very good CV levels suitable for screening [15]. This method has the advantages of high efficiency (multiple samples can be run in parallel), high accuracy, and full automation.
In this study, there was good correlation for measurements of HbA, HbF, and HbA2 between CE and CA. The percentage of agreement on Hb fractions between CE and CA was 89.5%. CA did not identify some HbA2 decreases and HbF increases. This may indicate that CE is more sensitive than CA for detecting Hb fractions. Because only one-third of globin gene mutations causing Hb abnormalities change the charge of Hb and one-quarter of the residues of Hb are internally situated, CA detects only 25% of hemoglobin variants [16, 17]. Several studies have revealed poor precision for HbA2 quantification methods based on electrophoresis [8]. In addition, the prominent feature of the CE method is the ability to separate HbA2 completely from HbE [10, 18]. Tandem mass spectrometry has been able to measure HbA2 in the presence of HbE. However, the present CE method provides this information in a simplified format [9].
CE patterns were easier to interpret than CA patterns. While the latter needs technical expertise, the former provides objective results and various settings available for interpretation, recording, and archives. However, the presence of HbA is essential for CE to obtain appropriate zones for verifying the identity of the Hb.
A practical method for routine Hb electrophoresis is cellulose acetate at alkaline pH. It is rapid and reproducible and separates Hbs A, F, S, C, and A2. The use of citrate agar electrophoresis at acid pH permits further separation of several common variants that migrate together on cellulose acetate: S from D and G, and C from E and O [19]. Nevertheless, citrate agar electrophoresis is not widely used because it is slow and difficult to standardize [20]. The agar plates must be freshly made and electrophoretic mobilities are often inconstant owing to the relatively high electrical resistance of the citrate agar, leading to variable effects on the Hb [20].
Various cell counter-based formulae have been published to investigate microcytosis and to discriminate between thalassemia and iron deficiency [21]. They are useful when resources are limited. However, none of the differentiation indices or formula provided 100% sensitivity and 100% specificity for discrimination purposes [21]. Further confirmatory tests are needed. Therefore, it is not suitable in Korea to screen patients with microcytosis using these formulae.
Migration of Hb variants is identified on 15 zones of the Minicap system. Zone 7 identifies HbF, as well as other Hb variants, including Hb Q-Thailand (G-Taichung), Hb Richmond, Hb G-San Jose, Hb Porto Alegre, Hb Presbyterian, Hb Alabama, Hb Chapel Hill, Hb Bassett, Hb Barcelona, Hb Geldrop Santa Anna, Hb Swan River, Hb Burke, Hb Manitoba I, Hb Manitoba II, "J-Paris-I" HbA2 variant, and denatured HbS. Therefore, when HbF levels are elevated, it has to be considered that other Hb variants, although scarce, are possible.
Reduced MCV, MCH, and HbA2 might indicate either iron deficiency or α-thalassemia [7]. In this study, 29 patients had decreased HbA2. Among these, 25 cases were diagnosed as IDA, and 4 cases were considered to have the α-thalassemia trait. This condition is benign and most patients are diagnosed on routine screening [22].
In the present study, one of the donors identified as having β-thalassemia was a Southeast Asian from Burma. This reflects the growing migration from areas with high prevalence of β-thalassemia to Korea and the importance of screening for pregnant women of those ethnic groups.
The β-thalassemia gene frequency in Koreans is approximately 0.1% or less [23]. In this study, the total incidence of abnormal hemoglobin fractions was 1.4% as detected by both CE and CA, which is slightly higher than the results previously reported [16]. We speculate that various factors, including the criteria used for selection of study samples, screening methods, and epidemiologic characteristics, may contribute to the different results of the studies. Screening of a larger population is needed to understand more precisely the incidence of hemoglobinopathies in the Korean population.
As the prevalence of hemoglobinopathies is very low in Korea, Hb electrophoresis has not been a routine test for evaluating anemia patients. However, underestimation of thalassemias and hemoglobinopathies may result in misdiagnosing anemic patients to IDA, thus causing the patients to suffer from prolonged inappropriate treatment. Furthermore, due to increased migration of ethnic minorities with high rates of globin gene defects to Korea, the problem of thalassemia has to be considered in this country. We suggest CE as a screening tool for hemoglobin disorders in the Korean population. CE is comparable to CA in terms of reliable measurement of Hb fractions, and is suitable for simple screening.
Go to :
 XML Download
XML Download