

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cystic Fibrosis (CF) is an autosomal recessive disease of exocrine tissues; the disease is characterized by the abnormal transport of ions and fluid across epithelial cell membranes. Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene are responsible for both classical and atypical presentations of the disease, including pulmonary disease, pancreatic insufficiency, malabsorption, meconium ileus, failure to thrive, infertility, and elevated concentrations of chloride in sweat [1]. A diagnosis of CF is based upon several findings, including the presence of typical clinical features, history of CF in a sibling, positive sweat test, identification of CFTR mutations in both alleles, and an abnormal nasal potential difference measurement [2].
The CFTR gene spans an approximately 240-kb region on chromosome 7q31.3 and consists of 27 exons [3-5]. It encodes a membrane protein of 1,480 amino acids that functions as a cAMP-regulated chloride channel in exocrine epithelia [6]. CF is a very common hereditary disease in Caucasian populations with incidences ranging from 1 in 2,500-3,500 in the U.S. [7, 8] and approximately 1 in 1,000-3,000 newborns in Europe [9]. However, CF is quite rare in Asian populations, and an epidemiological study in a Japanese population found the incidence of CF to be about 1 in 350,000 [10, 11]. CF is also extremely rare in the Korean population and only 10 patients with CF have been reported thus far, of which 7 cases were confirmed by genetic analysis [12-19].
There are currently over 1,700 mutations listed in the CFTR mutation database [20]. The most common mutation of the CFTR gene in Caucasian populations is a p.F508del, and this mutation is found in approximately 70% of Caucasian CF patients [1], whereas the incidence of other mutations remains about 2-3% per mutation [21]. Interestingly, the p.F508del is not a common mutation in Asian populations and has frequencies of 19-44% in Indians [22-25], 17.3% in Pakistanis [26], and 18.1% in Iranians [27]. Further, of the few reports of genomic DNA analysis of Japanese patients with CF, there have been no cases where a p.F508del mutation was found [11].
Currently, not much data is available on the spectrum of mutations in Korean CF patients. Here, we describe the cases of 2 CF patients and have summarized the mutational spectrum of the CFTR gene from Korean CF patients in order to further characterize the specific mutational pattern of the CFTR gene in the Korean population.
Go to : 
MATERIALS AND METHODS
1. Patients
Patient 1 was a 22-yr-old woman who was admitted for antibiotic therapy of infection with nontuberculous mycobacteria (NTM). The patient had been treated for several episodes of chronic productive cough, sputum, and pneumonia, which necessitated frequent visits to the outpatient clinic. At the age of 17, she was admitted to a civil hospital for the treatment of pulmonary tuberculosis and had receiv-ed antituberculosis drug therapy for 15 months. Her respiratory symptoms progressively worsened, and she was diagnosed with bronchiectasis and a NTM infection. She had an older brother with an unremarkable medical history. There was no evidence of exocrine pancreatic insufficiency or malabsorption. The sweat chloride concentration was measured by a quantitative pilocarpine iontophoresis sweat test, in accordance with the guidelines of the Clinical and Laboratory Standards Institute. The average sweat chloride concentration on both forearms was 81.2 mmol/L, which exceeded the upper limit of the reference range (<60 mmol/L).
Patient 2 was a 31-yr-old woman complaining of chronic productive cough, sputum, and recent hemoptysis without any gastrointestinal symptoms. The patient had been treated for these symptoms with antibiotics, but no improvement was noted. After a preliminary diagnosis of community-acquired pneumonia, she was admitted for further work-up and treatment. During her infancy, she had pertussis. She had recurrently received antituberculosis treatment from the age 15, with repeated events of remission and relapse of pulmonary tuberculosis. No evidence of exocrine pancreatic insufficiency or malabsorption was found. The average sweat chloride concentration was elevated (112.4 mmol/L), which was highly suggestive of CF.
2. Mutation analysis
Informed consent was obtained from all parents and family members, and patient confidentiality was been maintained. Genomic DNA was isolated from peripheral blood leukocytes by using the Wizard Genomic DNA Purification Kit, according to the manufacturer's instructions (Promega, Madison, WI, USA). The presence of mutations in all 27 exons and exon-intron boundaries of the CFTR gene was assessed by polymerase chain reaction (PCR) and direct sequencing using primer pairs designed by the authors. PCR was performed using a thermal cycler (Model 9600; Applied Biosystems, Foster City, CA, USA), and cycle sequencing was performed using the ABI Prism 3100 Genetic Analyzer with the BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems). Sequence variations were analyzed with reference to the wild-type sequence (GenBank Accession No. NM_00492.3) by using the Sequencher program (Gene Codes Corp., Ann Arbor, MI, USA).
When a large deletion mutation was thought to be present, multiplex ligation-dependent probe amplification (MLPA) analysis was performed using the SALSA P091-C1 CFTR test kit (MRC-Holland, Amsterdam, the Netherlands), according to the manufacturer's protocol [28]. The P091-C1 CFTR probemix contains 5 control fragments and 43 different probes with amplification products. Briefly, 100 ng of genomic DNA was denatured at 98℃ and hybridized with the CFTR-MLPA probe mix at 60℃ for 16 hr. Subsequently, the annealed probe was ligated to the Ligase-65 mix at 54℃ for 15 min. The appropriate PCR primers, dNTPs, SALSA PCR buffer, and SALSA polymerase were added, and the subsequent PCR reactions were performed. Each probe amplification product, 1 µL in volume, was then mixed with 0.5 µL GeneScan™500 ROX™ Size Standard (Applied Biosystems, Warrington, UK) and 8.5 µL HiDi Formamide (Applied Biosystems). Electrophoresis of the PCR products was performed using the ABI 3130 Genetic Analyzer (Applied Biosystems, Foster, CA, USA), and the MLPA data were analyzed using GeneMarker 1.6 software (SoftGenetics, LLC., State College, PA, USA). A normal copy number is expected to generate a normalized signal value of 1.
3. Clinical findings and spectrum of mutations in Korean CF patients
In order to study the spectrum of mutations in Korean CF patients, we reviewed the previously identified CF patients at our institution (N=4) [14, 15, 17, 18] and in the literature (N=3) [16, 19, 29] in addition to the cases analyzed in this report (N=2). Only genetically confirmed cases were included in this study.
Go to :
RESULTS
1. Genetic analysis of the 2 Korean patients
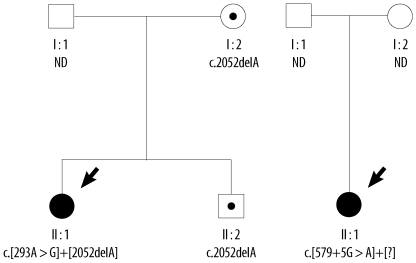
Two mutations in the CFTR gene were found in Patient 1: a heterozygous A to G transition in exon 4 [c.293A>G (p.Q98R)] and a heterozygous deletion of an A in exon 13 [c.2052delA (p.L728NfsX38)]. A family study showed that her mother and older brother both also possessed the c. 2052delA mutation. No targeted genetic testing was done on her father. Our search of the Cystic Fibrosis Mutation Database [20] showed that the p.Q98R and c.2052delA mutations have also been reported in CF patients from France.
In Patient 2, a heterozygous splicing mutation in intron 4 (c.579+5G>A) was identified, but we were unable to detect the mutation that must presumably have been present on the opposite allele. No further genetic study was done on her family members. The CFTR mutations from the families of the 2 patients are summarized in the pedigree (Fig. 1).
2. Clinical findings in 9 Korean CF patients
The average ages of 9 patients (7 female and 2 male patients) at diagnosis and at presentation of symptoms were 13 and 2.8 yr, respectively. Five patients were diagnosed with CF under the age of 16 and the other 4 patients were clinically diagnosed with CF during adulthood (Table 1). None of the patients had a history of sibling death due to respiratory or gastrointestinal problems or any symptoms related to CF in family members.
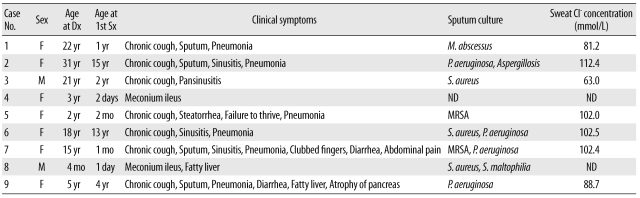
The respiratory symptoms were clinically documented in the CF patients as follows: chronic cough (N=7; 78%), sputum (N=4; 44%), sinusitis (N=4; 44%), recurrent or persistent pneumonia (N=7; 78%), and bronchiectasis (N=5; 56%). Infections related with CF were pulmonary tuberculosis (N=5; 56%), aspergillosis (N=1; 11%), and infections with NTM (N=1; 11%), Staphylococcus aureus (N=5; 56%), Pseudomonas aeruginosa (N=4; 44%), and Stenotrophomonas maltophilia (N=1; 11%). Most of the patients had experienced infections of S. aureus and P. aeruginosa, which are well-known clinical infections of CF, during the course of their disease progression. Other symptoms observed in the CF patients were a history of failure to thrive (N=1; 11%), steatorrhea (N=3; 33%), clubbed fingers (N=1; 11%), fatty liver (N=2; 22%), pancreatic atrophy (N=1, 11%) and meconium ileus (N=2; 22%) (Table 1).
All patients were diagnosed with CF on the basis of classical clinical phenotypes and high sweat chloride concentration (>60 mEq/L). The median sweat chloride concentration of 7 patients was 93.2 mmol/L, and all patients tested showed high sweat chloride concentration. In 2 neonates, the sweat test could not be done because of their young age.
3. Spectrum of mutations in the 9 Korean CF patients
A total of 16 disease-causing mutations of the CFTR gene were identified in the 9 Korean CF patients (Table 2). All identified mutations were detected by PCR and direct sequencing with the exception of a large deletion in exon 14a, which was detected by MLPA. No p.F508del mutations, known to be the most common mutation among Caucasians, were detected in the Korean patients of this study. The identified mutations included 3 missense mutations (p.Q98R, p.Q1352H, and p.L441P), 3 nonsense mutations (p.Q220X, p.Q1291X, and p.L88X), 1 duplication with fra-meshift (c.3908dupA), 1 insertion with frameshift (c.2089-2090insA), 4 splice site mutations (c.1766+2T>C, c.3272-26A>G, c.579+5G>A, and IVS8-T5) and 2 deletion mutations (c.2052delA and c.2623-?_2751+?del).
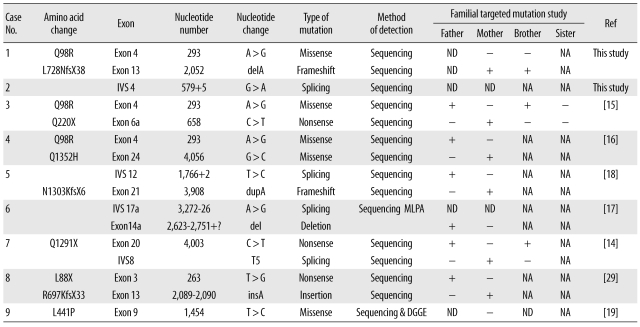
The p.Q98R mutation was the only recurrently observed mutation, with a frequency of 18.8% (3/16 alleles). Mutations on both alleles were identified in 7 out of 9 patients. All identified mutations of the CF patients examined were confirmed with targeted genetic tests in consenting family members. Eight out of 9 families were tested, and all parents of the patients, except in 1 family (case 9), were proven to be heterozygous carriers of the mutations with no phenotypic abnormalities (Table 2).
Go to :
DISCUSSION
Accurate knowledge of CF mutations in specific populations provides information for CF prevention programs applicable via heterozygote screening and prenatal diagnosis [25]. The identification of mutations and their frequencies is thus critically important for designing gene probes for effective diagnosis of CF in a given population. Further, different mutations are associated with varying severity of disease with implications on management and prognosis [26]. However, limited data are available on the spectrum of mutations in Korean CF patients. Therefore, this study was planned to characterize the mutational spectrum of the CFTR gene in Korean CF patients.
Currently over 1,700 mutations are listed in the Cystic Fibrosis Mutation Database [20]. However, only a few mutations have been reported in the Korean population. No mutation, except p.Q98R, has repeatedly been observed in Korean CF patients in this study. Furthermore, only one of the 32 common mutations found in the screening panel for Caucasians was identified in this Korean population, namely, the c.3272-26A>G mutation [20].
A noteworthy characteristic of the mutational spectrum of the Korean CFTR gene is the absence of mutations that are common in the European and American populations. This highly heterogeneous mutational spectrum suggests that a screening set of common CFTR mutations is of little use for the Korean population. Rather, sequencing of entire gene and complementary MLPA might be a more feasible way to accurately confirm a diagnosis of CF. Sequencing analysis detects more than 98% of CFTR mutations [30], and MLPA can detect deletions not identified by sequence analysis. However, the mutation detection rate of MLPA is not currently known.
Although CF tends to be detected early in life, diagnosis of CF is being made in adults with increased frequency [31, 32]. Patients who are diagnosed with CF when they are adults normally present with chronic respiratory problems. As a group, they have a milder variant of lung disease, a lesser degree of Pseudomonas infection, and are more likely to be pancreatic-sufficient than patients in whom CF is diagnosed at earlier ages [31, 32]. In the Korean CF patients in our study, half were diagnosed when they were adults and presented with chronic respiratory problems such as chronic productive cough, sputum, and recurrent pneumonia and presented with bronchiectasis in a more advanced disease. Pseudomonal and staphylococcal infections were observed in 4 (4/9, 44%) and 5 (5/9, 56%) patients, respectively. Further, the lower number of reported CF diagnoses in the Korean population may suggest that CF is under-diagnosed in Korea.
In Patients 2 and 9, one of the causative mutations was not identified either by direct sequencing or by MLPA analysis. The CFTR mutation detection rate varies depending on the test method and ethnic background. In some symptomatic individuals, only one or neither disease-causing mutation is detectable; in some carriers, the disease-causing mutation is not detectable. This may indicate a molecular defect in a non-coding or promoter region or a deep intron-ic region of the gene, which may have led to a truncated or abnormal protein as a result of deregulated promoter signals or abnormal splicing. Moreover, the splicing mutation observed in Patient 2 (c.579+5G>A) has also been reported in CF patients from Italy, according to the Cystic Fibrosis Mutation Database [20].
Physicians who are not familiar with the spectrum of CF's phenotypic manifestations may not consider CF when performing their differential diagnoses, thus delaying accurate diagnosis [15]. As a result, CF patients are frequently misdiagnosed and treated for tuberculosis and other diseases. Five CF patients (56%) from our study had previously been diagnosed and treated for pulmonary tuberculosis. Although sweat chloride analysis is considered to be the gold standard for screening CF, the test is difficult to perform because of insufficient standardization of the procedure and issues with reimbursement in Korea.
Whilst the frequency of the p.F508del mutation is 87% in Northern Europe, it is only 28% in Asia and the Middle East, with a combined worldwide frequency of 66% (The Cystic Fibrosis Mutation Database 2010; The CF Genetic Analysis Consortium [1994]) [26]. No p.F508del mutations were detected in the Korean patients of this study. CF mutation frequencies vary widely among different populations, and haplotype heterogeneity has been found to be greater in populations with lower p.F508del frequencies [33]. The heterogeneous mutational spectrum observed in this study also confirms these previous findings of high haplotype heterogeneity with lower p.F508del frequencies. Therefore, screening for CF with p.F508del mutation analysis alone is not recommended in Korean patients.
In summary, we report the cases of 2 Korean CF patients with 3 known CFTR mutations (p.Q98R, c.2052delA, and c.579+5G>A), and these mutations have been submitted to the Cystic Fibrosis Mutation Database [20]. The heterogeneous mutational spectrum of the CFTR gene in this Korean population suggests that full sequencing, covering a broader range of the CFTR genome, is a better diagnostic method for Korean CF patients. In addition, MLPA analysis for the detection of large deletions or duplications can be used as a complementary tool to the direct sequencing method. Even though a large amount of research is required before all the CFTR mutations in the Korean population are characterized by whole gene sequencing, future genomics tools will allow us to develop more advanced diagnostic era for cystic fibrosis. This can be made possible by standardized molecular tests utilizing whole genome sequencing and full gene sequencing accompanied by MLPA.
Go to :
 XML Download
XML Download