INTRODUCTION
Candida albicans is the most common species causing
Candida bloodstream infections (BSI). Nosocomial
C. albicans BSI develop primarily as a consequence of endogenous colonization, but are often caused by a strain acquired in hospital settings [
1,
2]. Therefore, to identify the source of BSI caused by
C. albicans, a suitable strain typing method is essential. In recent years, genomic microevolution (also called microvariation) has been documented for multiple
C. albicans isolates from blood and other anatomic sites in individual patients with
Candida BSI [
1,
3-
6]. Pfaller et al. [
5] demonstrated that
C. albicans BSI strains that are endemic in some hospitals undergo microevolution in the hospital setting. Given the rapid and frequent occurrence of
C. albicans microevolution, assessing genetic relatedness among
C. albicans isolates from patients with BSI is particularly important for understanding and controlling nosocomial
C. albicans BSI within hospitals.
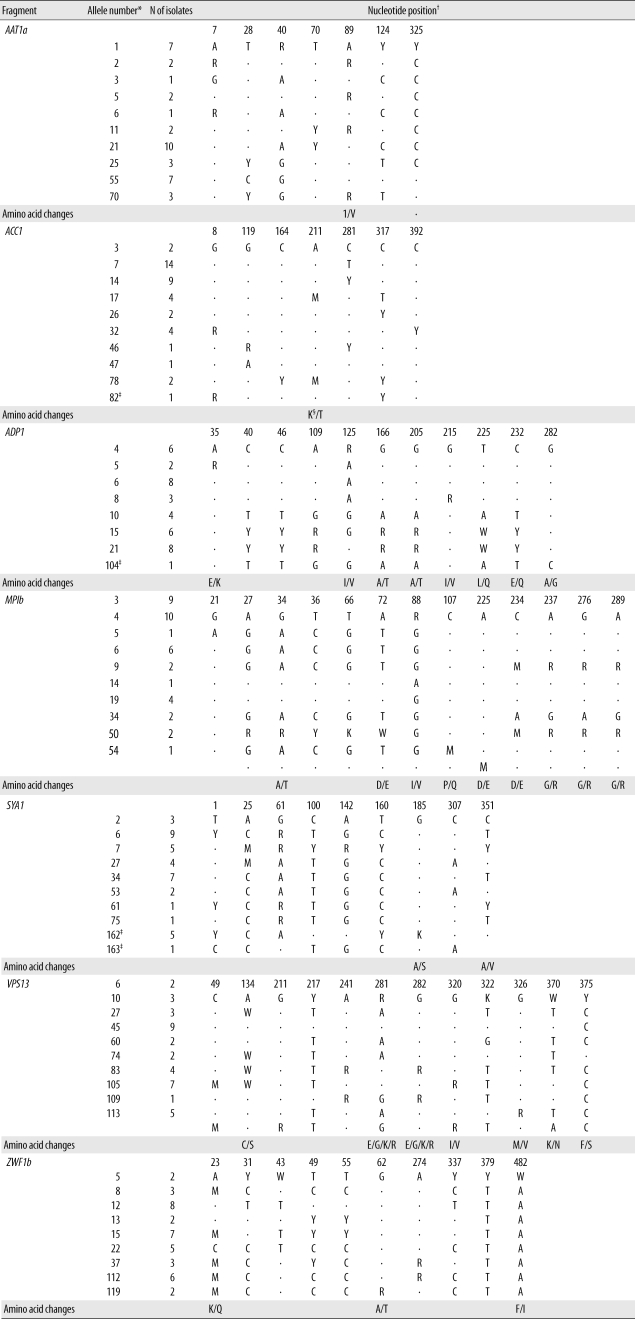
Multilocus sequence typing (MLST), which is based on DNA sequence analysis of nucleotide polymorphisms within housekeeping gene fragments, is a reliable, objective, and highly discriminating molecular typing method for the characterization of clinical
C. albicans isolates [
6,
7]. In addition, MLST permits the exchange of molecular typing data via the internet, which facilitates both local and global epidemiologic studies [
7,
8]. To date, simultaneous comparisons of pulsed-field gel electrophoresis (PFGE), Southern blot hybridization, and MLST methods for genotyping
C. albicans isolates from BSI patients has not been performed.
By using the MLST method, we analyzed the genetic relationships among C. albicans isolates from blood cultures and other anatomical sites from 21 patients with candidemia admitted to the same hospital over a 3-yr period. The purpose of this study was to evaluate the usefulness of MLST relative to PFGE and Southern blot hybridization fingerprinting tools for detecting nosocomial clusters vs. microevolution among C. albicans isolates from patients with C. albicans BSI.
Go to :

DISCUSSION
MLST allows the exchange of genotyping data via the internet for global epidemiology [
7,
8]. A large amount of MLST data of
C. albicans has accumulated from diverse locations, and it has been analyzed on a website. To date, an MLST study of
C. albicans isolates has not been performed in Korea. The present study showed that some
C. albicans strains that cause candidemia at our hospital shared the same MLST type with strains from other geographic regions of the world. Overall, 22 isolates from 12 patients belonged to new sequence types (10 DSTs), whereas 23 isolates from 14 patients belonged to previously described types (10 DSTs). In this study, 4 polymorphic nucleotide sites were identified. Our study showed that MLST allows an exchange of international genotyping data and offers distinctive standardization and portability.
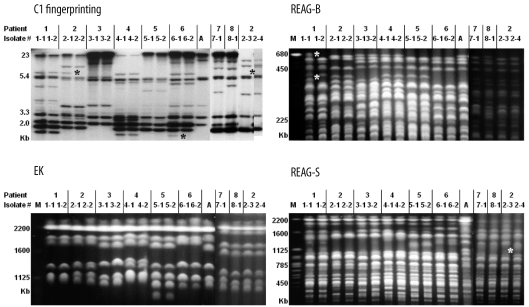
Chen et al. [
8] reported that MLST is superior to REAG-B for tracing the microevolution of
C. albicans strains within the same patient. However, simultaneous comparison of MLST, REAG-B, REAG-S, and C1 fingerprinting techniques for the detection of microevolution of
C. albicans strains from patients with BSI has not been performed. In this study, multiple isolates from the same patient showing different DSTs were examined. By MLST, 1 or 2 allelic differences, indicative of microevolution [
7], were found in 6 sets of isolates. REAG-B, REAG-S, and C1 fingerprinting also detected microevolution among 5, 2, and 5 sets of isolates from the same patient. However, none of these 4 methods detected all 7 sets of microevolution that were detected by combining all 4 methods used in this study. These results show that MLST is as efficient as C1 fingerprinting and REAG for detecting microevolution among sequential
C. albicans isolates from
Candida BSI.
Two previous studies showed that MLST analysis has the highest levels of concordance with Ca3 Southern hybridization [
10] and REAG-B in discriminating epidemiologically related strains [
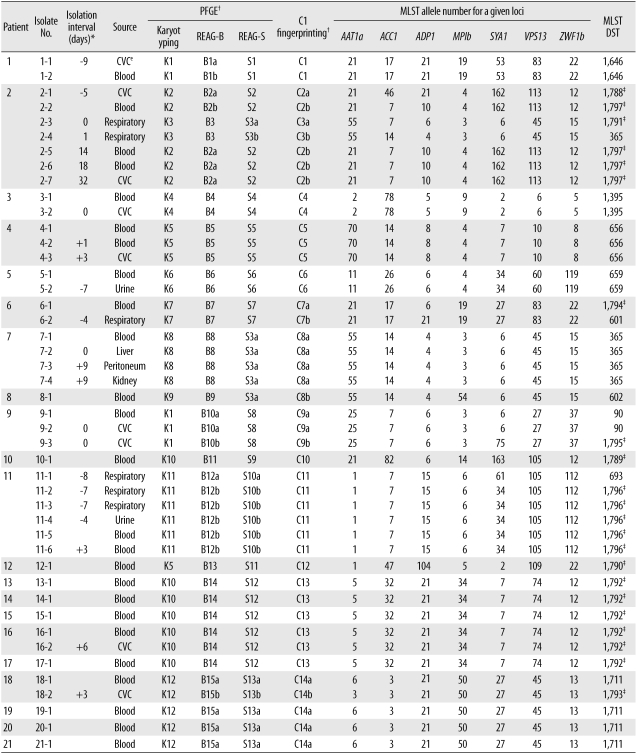
8]. We analyzed 2 previously recognized nosocomial clusters of BSI that had been evaluated by PFGE and C1 fingerprinting techniques with MLST. MLST analysis clearly identified 9 patients' isolates from 2 nosocomial clusters as 2 DSTs (DSTs 1711 and 1792). Of these 9 patient isolates, 1 catheter isolate from patient 18 showed a different DST (DST 1793) from corresponding blood isolates (DST 1711); however, it differed by only 1 locus, suggesting microevolution. This finding, together with microevolution observed among serial isolates from the same patient, suggest that closely related
C. albicans strains, which have different DSTs only at 1 or 2 of the sequenced alleles, may be classified as epidemiologically related strains.
Of particular note in our study is that 1 set of isolates classified as the same DST (DST 365) by MLST differed in PFGE and C1 fingerprinting patterns. In addition, 3 sets of isolates from different patients in whom MLST showed 1 or 2 allelic differences (closely related), also revealed different patterns by PFGE or C1 fingerprinting. These isolates from 4 sets, which showed the same or similar MLST but exhibited different PFGE or C1 fingerprinting patterns, were obtained from 3 epidemiologically unrelated patients. These results suggest that MLST may be less useful for long-term epidemiological surveillance of C. albicans isolates in a hospital setting, and that REAG-B perhaps constitutes the more useful method for confirming MLST findings. In addition, our data show that MLST analysis of C. albicans isolates, in the absence of epidemiologic data, may not be recommended as a routine component of infection control, but that it may be useful in evaluating putative outbreaks identified by conventional hospital surveillance.
Odds et al. [
11] also noted a few examples among their isolates that were very closely related according to MLST analysis, but that differed in ABC type, which is a very stable epidemiological marker in
C. albicans isolates. Our results, together with those by Odds et al. [
11], highlight the limitation of the MLST analysis in typing diverse strains of
C. albicans from different patients (inter-patient analysis). These data suggest that frequent genetic evolutionary changes in
C. albicans isolates may affect the interpretation of MLST data, especially for detecting inter- and intra-hospital spread of
C. albicans strains. Therefore, the
C. albicans strains at our hospital that shared the same DST with strains from other geographic regions of the world need further characterizing by PFGE or C1 fingerprinting.
In summary, we compared MLST, PFGE, and C1 fingerprinting for typing C. albicans isolates from patients with BSI. The results show that although MLST is a powerful tool for discriminating and detecting microevolution among C. albicans isolates, it has some limitations in typing diverse strains of C. albicans from different patients. Therefore, we suggest that REAG or C1 fingerprinting can be a very helpful confirmatory tool in instances when C. albicans isolates from different patients show the same or similar (differ by only 1 or 2 alleles) MLST types.
Go to :


 PDF
PDF ePub
ePub Citation
Citation Print
Print


 XML Download
XML Download