

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
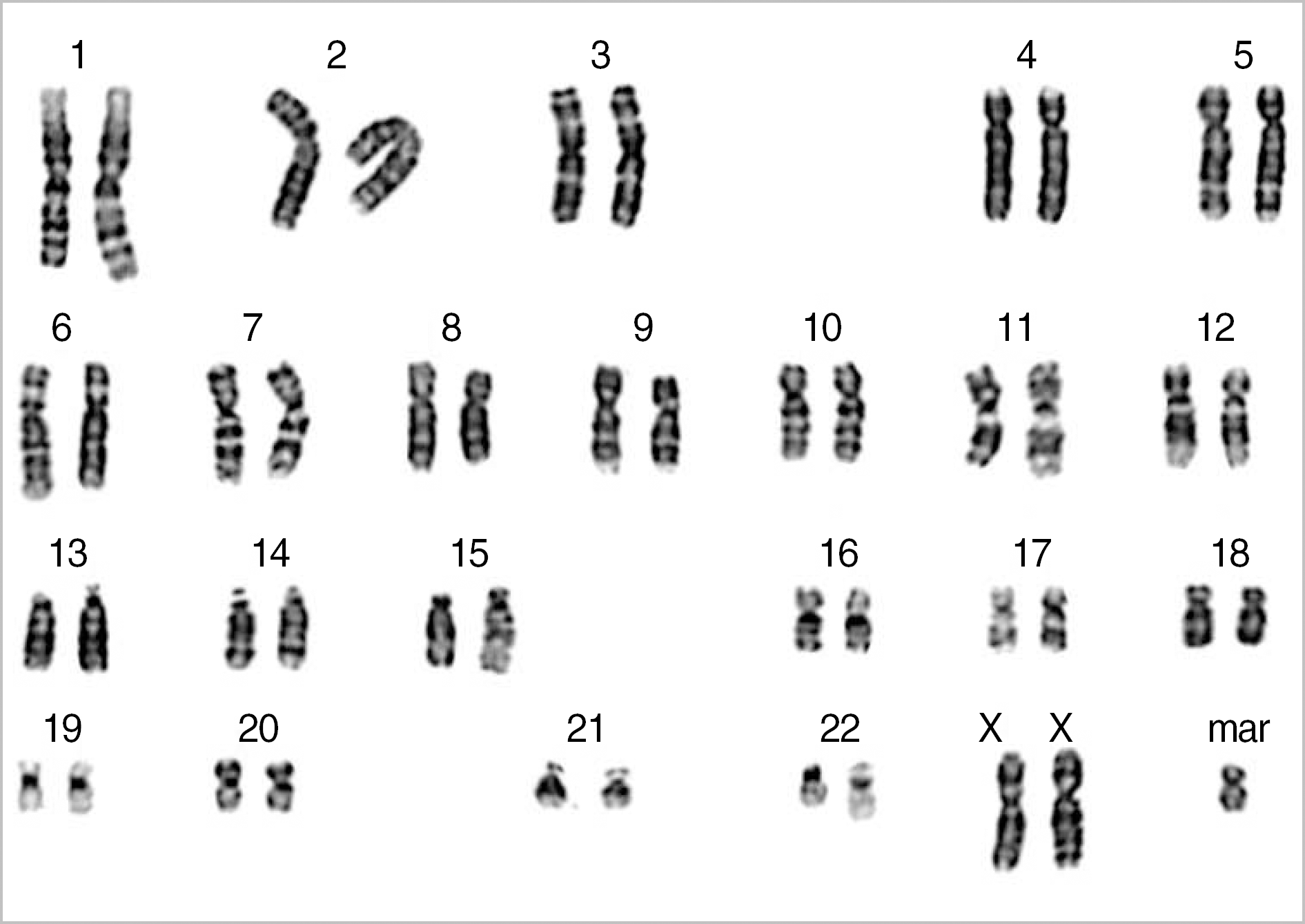
Partial trisomy 2p is a rare but relatively well-defined syndrome with distinctive clinical features, including marked psychomotor delay, dysmorphic face, and congenital heart disease. The phenotype of trisomy 18p is variable, from normal appearance to moderate mental retardation. Most cases of trisomy 2p and trisomy 18p result from the inheritance of an unbalanced segregant from a balanced parental translocation or due to de novo duplication. Here, we present the first report of a combined partial trisomy 2p and trisomy 18p due to a supernumerary marker chromosome (SMC). The final karyotype of the patient was 47,XX,+der(18)t(2;18)(p23.1;q11.1)[22]/46,XX[8]. The patient had typical dysmorphic features of partial trisomy 2p23-pter syndrome and congenital heart disease. SMCs are remarkably variable in euchromatic DNA content and mosaicism level. The precise identification of the origin and composition of SMCs is essential for genotype-phenotype correlation and genetic counseling.
Go to : 
REFERENCES
1.Shaffer LG., Slovak ML, et alISCN 2009: an international system for human cytogenetic nomenclature (2009). Basel: Karger. 2009. 73–4.
2.Viersbach R., Engels H., Schwanitz G. Identification of supernumerary der(20) chromosomes by FISH in three patients. Am J Med Genet. 1997. 70:278–83.

3.Douet-Guilbert N., Marical H., Pinson L., Herry A., Le Bris MJ., Morel F, et al. Characterisation of supernumerary chromosomal markers: a study of 13 cases. Cytogenet Genome Res. 2007. 116:18–23.
4.Liehr T., Mrasek K., Weise A., Dufke A., Rodriguez L., Martinez Guardia N, et al. Small supernumerary marker chromosomes—progress towards a genotype-phenotype correlation. Cytogenet Genome Res. 2006. 112:23–34.
5.Starke H., Nietzel A., Weise A., Heller A., Mrasek K., Belitz B, et al. Small supernumerary marker chromosomes (SMCs): genotypephenotype correlation and classification. Hum Genet. 2003. 114:51–67.
6.Cho BH., Park HW. The standardization study of Korean Bayley Scales of Infant Development (K-BSID-II): analyses of Korean infants performance of K-BSID-II in terms of demographical variables. Korean J Dev Psychol. 2004. 17:191–206.
7.Shaffer LG., Slovak ML, et alISCN 2009: an international system for human cytogenetic nomenclature (2009). Basel: Karger. 2009. 105–28.
8.Francke U., Jones KL. The 2p partial trisomy syndrome. Duplication of region 2p23 leads to 2pter in two members of a t(2;7) translocation kindred. Am J Dis Child. 1976. 130:1244–9.
9.Gruchy N., Jacquemont ML., Lyonnet S., Labrune P., El Kamel I., Siffroi JP, et al. Recurrent inverted duplication of 2p with terminal deletion in a patient with the classical phenotype of trisomy 2p23-pter. Am J Med Genet A. 2007. 143A:2417–22.
10.Lurie IW., Ilyina HG., Gurevich DB., Rumyantseva NV., Naumchik IV., Castellan C, et al. Trisomy 2p: analysis of unusual phenotypic findings. Am J Med Genet. 1995. 55:229–36.
11.Roggenbuck JA., Fink JM., Mendelsohn NJ. Unique case of trisomy 2p24.3-pter with no associated monosomy. Am J Med Genet. 2001. 101:50–4.
12.Al-Saffar M., Lemyre E., Koenekoop R., Duncan AM., Der Kaloustian VM. Phenotype of a patient with pure partial trisomy 2p(p23→pter). Am J Med Genet. 2000. 94:428–32.
13.Roberts AE., Irons MB., Kimonis VE., Mulliken JB., Morton CC., Lee C, et al. Description of a case of distal 2p trisomy by array-based comparative genomic hybridization: a high resolution genome-wide investigation for chromosomal aneuploidy in a single assay. Am J Med Genet A. 2004. 130A:204–7.
14.Sock E., Rettig SD., Enderich J., Bosl MR., Tamm ER., Wegner M. Gene targeting reveals a widespread role for the high-mobility-group transcription factor Sox11 in tissue remodeling. Mol Cell Biol. 2004. 24:6635–44.
15.Romm E., Nielsen JA., Kim JG., Hudson LD. Myt1 family recruits histone deacetylase to regulate neural transcription. J Neurochem. 2005. 93:1444–53.
16.Iwahara T., Fujimoto J., Wen D., Cupples R., Bucay N., Arakawa T, et al. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. 1997. 14:439–49.
17.Lo-Castro A., Giana G., Fichera M., Castiglia L., Grillo L., Musumeci SA, et al. Deletion 2p25.2: a cryptic chromosome abnormality in a patient with autism and mental retardation detected using aCGH. Eur J Med Genet. 2009. 52:67–70.
18.Vrijenhoek T., Buizer-Voskamp JE., van der Stelt I., Strengman E., Sabatti C., Geurts van Kessel A, et al. Recurrent CNVs disrupt three candidate genes in schizophrenia patients. Am J Hum Genet. 2008. 83:504–10.
19.Dowa Y., Yamamoto T., Abe Y., Kobayashi M., Hoshino R., Tanaka K, et al. Congenital neuroblastoma in a patient with partial trisomy of 2p. J Pediatr Hematol Oncol. 2006. 28:379–82.
20.Rodriguez L., Liehr T., Mrasek K., Mansilla E., Martinez-Fernandez ML., Garcia A, et al. Small supernumerary chromosome marker generating complete and pure trisomy 18p, characterized by molecular cytogenetic techniques and review. Am J Med Genet A. 2007. 143A:2727–32.
21.Moog U., Engelen JJ., de Die-Smulders CE., Albrechts JC., Loneus WH., Haagen AA, et al. Partial trisomy of the short arm of chromosome 18 due to inversion duplication and direct duplication. Clin Genet. 1994. 46:423–9.
22.Marical H., Le Bris MJ., Douet-Guilbert N., Parent P., Descourt JP., Morel F, et al. 18p trisomy: a case of direct 18p duplication characterized by molecular cytogenetic analysis. Am J Med Genet A. 2007. 143A:2192–5.
23.Swingle HM., Ringdahl J., Mraz R., Patil S., Keppler-Noreuil K. Behavioral management of a long-term survivor with tetrasomy 18p. Am J Med Genet A. 2006. 140:276–80.
24.Crolla JA., Youings SA., Ennis S., Jacobs PA. Supernumerary marker chromosomes in man: parental origin, mosaicism and maternal age revisited. Eur J Hum Genet. 2005. 13:154–60.
25.Brondum-Nielsen K., Mikkelsen M. A 10-year survey, 1980-1990, of prenatally diagnosed small supernumerary marker chromosomes, identified by FISH analysis. Outcome and follow-up of 14 cases diagnosed in a series of 12,699 prenatal samples. Prenat Diagn. 1995. 15:615–9.
26.Gravholt CH., Friedrich U. Molecular cytogenetic study of supernumerary marker chromosomes in an unselected group of children. Am J Med Genet. 1995. 56:106–11.
27.Graf MD., Christ L., Mascarello JT., Mowrey P., Pettenati M., Stetten G, et al. Redefining the risks of prenatally ascertained supernumerary marker chromosomes: a collaborative study. J Med Genet. 2006. 43:660–4.
28.Shin YB., Nam SO., Seo EJ., Kim HH., Chang CL., Lee EY, et al. Partial trisomy 1q41 syndrome delineated by whole genomic array comparative genome hybridization. J Korean Med Sci. 2008. 23:1097–101.
Go to :
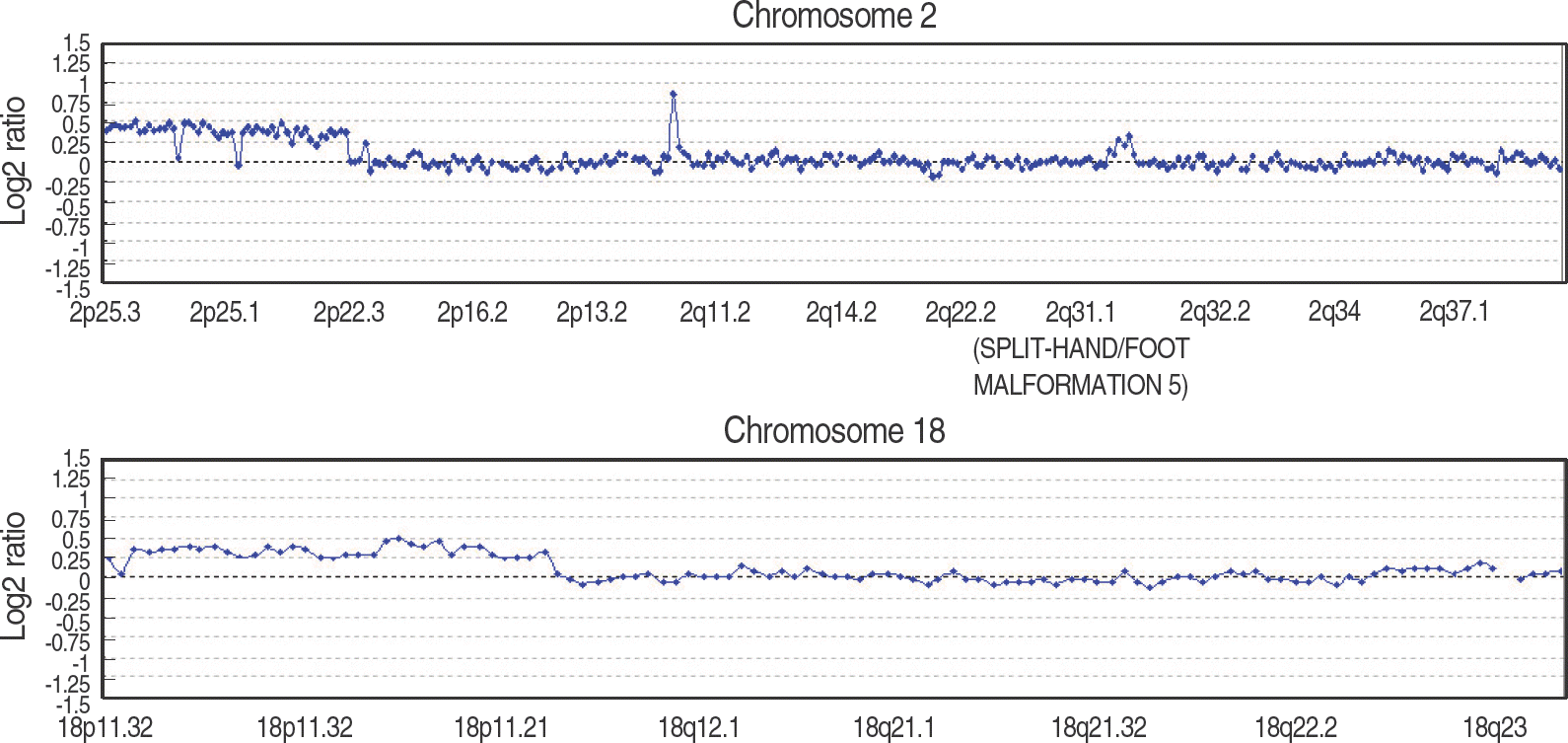 | Fig. 2.The ratio plots from array comparative genomic hybridization (aCGH) for chromosomes 2 and 18. The test sample was labeled with cyanine 3, and the reference sample was labeled with cyanine 5. Log2 ratio was calculated as log2(cy3)-log2(cy5), and a gain of particular clone is manifested as an upward deviation from the modal value 0.25. Gain of 46 clones on 2p and 30 clones on 18p were observed. The result of aCGH revealed arr 2p25.3p23.1(18,179-21,517,981)×3,18p11.32-18q11.1(59,690-17,039,831)×3. |
 | Fig. 3.FISH using whole chromosome painting probes (×1,000) of chromosomes 2 (labeled with Texas Red) and 18 (labeled with FITC) showed 2 signals of chromosomes 2 (red arrows), 2 signals of chromosome 18 (green arrows), and a small derivative chromosome comprising chromosomes 2 and 18 (yellow arrowhead). |
Table 1.
Main clinical features in patients with partial trisomy 2p23-pter syndrome
 XML Download
XML Download