

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
Background
Myotonic dystrophy type 1 (DM1) is an autosomal-dominant muscular dystrophy caused by expansion of cytosine-thymine-guanine (CTG) trinucleotide repeats in the myotonic dystrophy protein kinase (DMPK) gene. The clinical features of DM1 are multisystemic and highly variable, and the unstable nature of CTG expansion causes wide genotypic and phenotypic presentations. The aim of this study was to characterize the molecular and clinical spectra of DM1 in Koreans.
Methods
The CTG repeats of 283 Korean individuals were tested by PCR fragment analysis and Southern blot. The following characteristics were assessed retrospectively: spectrum of CTG expansions, clinical findings, genotype-phenotype correlation, anticipation, and genetic instability.
Results
One-hundred twenty-four patients were confirmed as DM1 by molecular tests, and the CTG expansions ranged from 50 to 2,770 repeats (median 480 repeats). The most frequent clinical features were myotonia, muscular weakness, and family history. Patients with muscular weakness or dysfunction of the central nervous system harbored larger CTG expansions than those without each symptom (P<0.05). The age of onset was inversely correlated with the size of the CTG expansion (γ=-0.422, P<0.001). The instability of CTG expansion representing as the maximum difference between sibships was observed from 50 to 700 repeats in nine families. Clinical anticipation and the increase in CTG repeat were significantly higher in maternally transmitted alleles (P=0.002).
REFERENCES
1.Kurihara T. New classification and treatment for myotonic disorders. Intern Med. 2005. 44:1027–32.

2.Brook JD., McCurrach ME., Harley HG., Buckler AJ., Church D., Aburatani H, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992. 68:799–808.
3.Machuca-Tzili L., Brook D., Hilton-Jones D. Clinical and molecular aspects of the myotonic dystrophies: a review. Muscle Nerve. 2005. 32:1–18.
4.Day JW., Ranum LP. RNA pathogenesis of the myotonic dystrophies. Neuromuscul Disord. 2005. 15:5–16.
5.Harper PS, editor. Myotonic dystrophy. 3rd ed.London: WB Saunders;2001. p. 47–86. p. 324–49.
6.The International Myotonic Dystrophy Consortium (IDMC). New nomenclature and DNA testing guidelines for myotonic dystrophy type 1 (DM1). Neurology. 2000. 54:1218–21.
7.Deka R., Majumder PP., Shriver MD., Stivers DN., Zhong Y., Yu LM, et al. Distribution and evolution of CTG repeats at the myotonin protein kinase gene in human populations. Genome Res. 1996. 6:142–54.
8.Martorell L., Monckton DG., Sanchez A., Lopez De Munain A., Baiget M. Frequency and stability of the myotonic dystrophy type 1 premutation. Neurology. 2001. 56:328–35.
9.Shim SH., Cho YH., Choi SK., Chung SR. The CTG repeat polymorphisms of myotonic dystrophy (DM) gene in Korean population. J Genet Med. 1997. 1:23–6.
10.Choi BO., SunWoo IN., Kim SM., Lee JS., Lee EK., Park KD, et al. Clinical significance of CTG repeat expansion in Korean myotonic dystrophy patients. J Korean Neurol Assoc. 1999. 17:548–53. (최병옥, 선우일남, 김승민, 이진성, 이은경, 박기덕 등. 한국인 근긴장성 이양증에서CTG 반복확장의임상적의의. 대한신경과학회지 1999;17: 548-53.).
11.Shojasaffar B., Moradin N., Kahrizi K., Cobo AM., Najmabadi H. CTG expansion & haplotype analysis in DM1 gene in healthy Iranian population. Can J Neurol Sci. 2008. 35:216–9.
12.Pan H., Lin HM., Ku WY., Li TC., Li SY., Lin CC, et al. Haplotype analysis of the myotonic dystrophy type 1 (DM1) locus in Taiwan: implications for low prevalence and founder mutations of Taiwanese myotonic dystrophy type 1. Eur J Hum Genet. 2001. 9:638–41.
13.Tishkoff SA., Goldman A., Calafell F., Speed WC., Deinard AS., Bonne-Tamir B, et al. A global haplotype analysis of the myotonic dystrophy locus: implications for the evolution of modern humans and for the origin of myotonic dystrophy mutations. Am J Hum Genet. 1998. 62:1389–402.
14.Culjkovic B., Stojkovic O., Vukosavic S., Savic D., Rakocevic V., Apostolski S, et al. CTG repeat polymorphism in DMPK gene in healthy Yugoslav population. Acta Neurol Scand. 2002. 105:55–8.
15.Marchini C., Lonigro R., Verriello L., Pellizzari L., Bergonzi P., Damante G. Correlations between individual clinical manifestations and CTG repeat amplification in myotonic dystrophy. Clin Genet. 2000. 57:74–82.
16.Gharehbaghi-Schnell EB., Finsterer J., Korschineck I., Mamoli B., Binder BR. Genotype-phenotype correlation in myotonic dystrophy. Clin Genet. 1998. 53:20–6.
17.Savic D., Rakocvic-Stojanovic V., Keckarevic D., Culjkovic B., Stojkovic O., Mladenovic J, et al. 250 CTG repeats in DMPK is a threshold for correlation of expansion size and age at onset of juvenile-adult DM1. Hum Mutat. 2002. 19:131–9.
18.Brisson D., Tremblay M., Prevost C., Laberge C., Puymirat J., Mathieu J. Sibship stability of genotype and phenotype in myotonic dystrophy. Clin Genet. 2002. 62:220–5.
19.Ashizawa T., Anvret M., Baiget M., Barcelo JM., Brunner H., Cobo AM, et al. Characteristics of intergenerational contractions of the CTG repeat in myotonic dystrophy. Am J Hum Genet. 1994. 54:414–23.
20.Amiel J., Raclin V., Jouannic JM., Morichon N., Hoffman-Radvanyi H., Dommergues M, et al. Trinucleotide repeat contraction: a pitfall in prenatal diagnosis of myotonic dystrophy. J Med Genet. 2001. 38:850–2.
21.Harley HG., Rundle SA., MacMillan JC., Myring J., Brook JD., Crow S, et al. Size of the unstable CTG repeat sequence in relation to phenotype and parental transmission in myotonic dystrophy. Am J Hum Genet. 1993. 52:1164–74.
Fig. 1.
The schematic map of the DMPK gene with relative location of CTG repeats, exons near the repeats, primers, and probe used in this study. The CTG repeats are detected by PCR using primers 102-F and 101-R when the fragment is less than 500 bp. To detect large expansions of more than 500 bp, genomic DNA was digested with the restriction endonucleases EcoRI or PstI. The EcoRI digestion results in ~9 kb band without Alu element, or ~10 kb band with Alu insertion. The PstI digestion results in ~1.2 kb and ~0.8 kb bands. Probe pM10M-6 is ~1.4 kb fragment containing the CTG repeats. Each fragment size is from the allele with Alu insertion and (CTG)20 repeats.
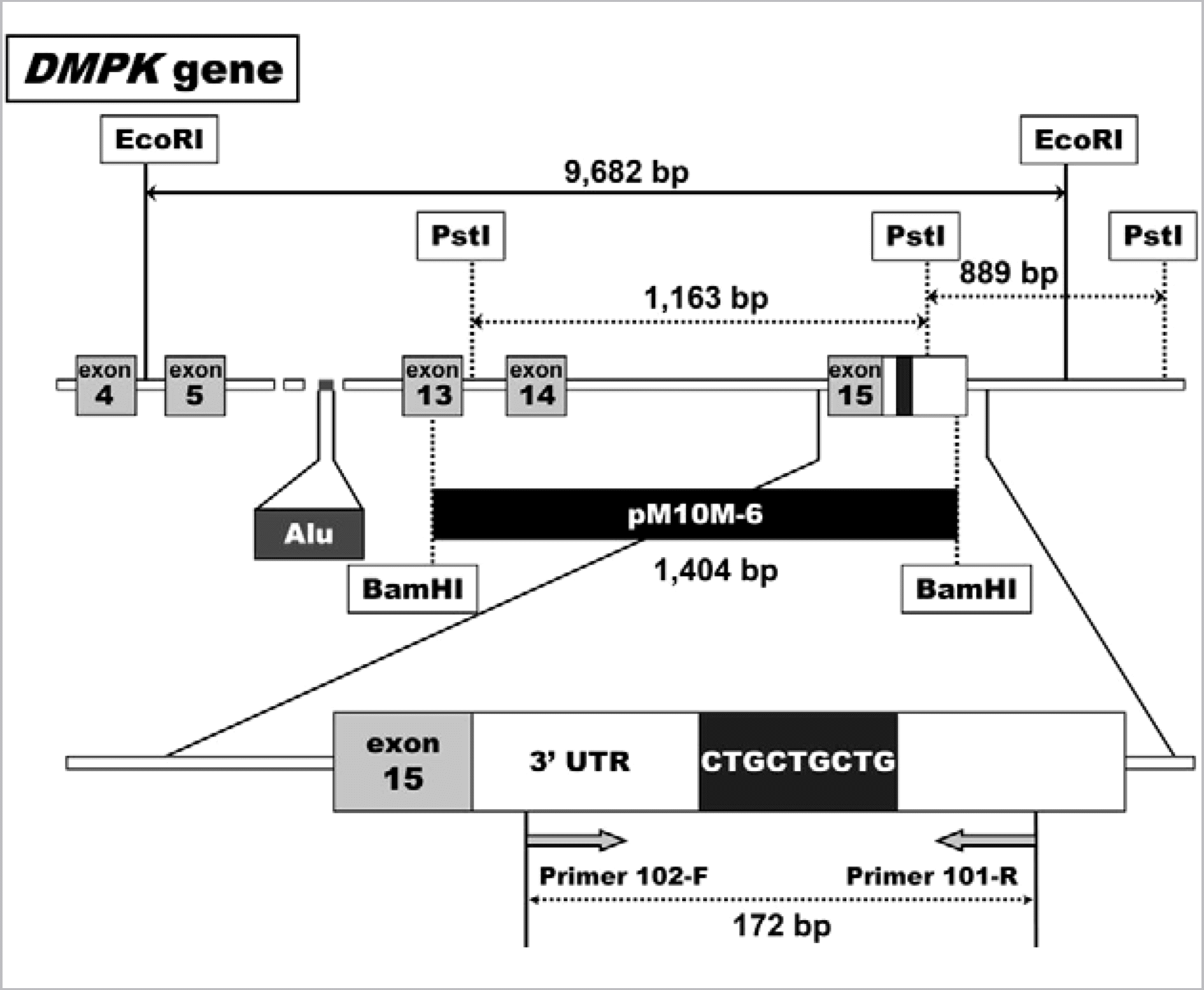
Fig. 2.
Representative results from (A) PCR and fragment analysis and (B) Southern blot. (A) Chromatogram obtained (a) from a normal individual with heterozygous 12 and 17 CTG re- peats. (b) from a normal individual with homozygous five CTG repeats. (c) from a DM1 patient with one normal 13 CTG repeat in the upper panel and the expanded 129 repeats in the lower panel. The chromatogram in lower panel was obtained after longer time electrophoresis. (B) (a) an autoradiogram obtained from four individuals by Southern blot after EcoRI digestion, and (b) after PstI digestion. In autoradiogram (a), a normal allele of ~9 kb was observed in lanes 1, 2, and 4. Expanded mutant allele larger than ~10 kb was observed in lanes 1 and 2 (marked by asterisk). The expansion of CTG repeats was also suspected in lane 3 (marked by arrow), but not clearly discriminated from a normal band of ~10 kb length. In autoradiogram (b), two normal bands of ~0.8 kb and ~1.2 kb were observed in each lane, and an additional expanded band was observed in lanes 1, 2, and 3. The expanded alleles were estimated as ~690 repeats in lane 1 and ~980 repeats in lane 2. An expanded allele of 75 repeats observed in lane 3 was also determined by PCR and fragment analysis (data not shown). Therefore, these three patients were diagnosed as DM1. Lane 4 was an unaffected individual, heterozygous for 6 and 12 CTG repeats (data from PCR and fragment analysis, not shown).
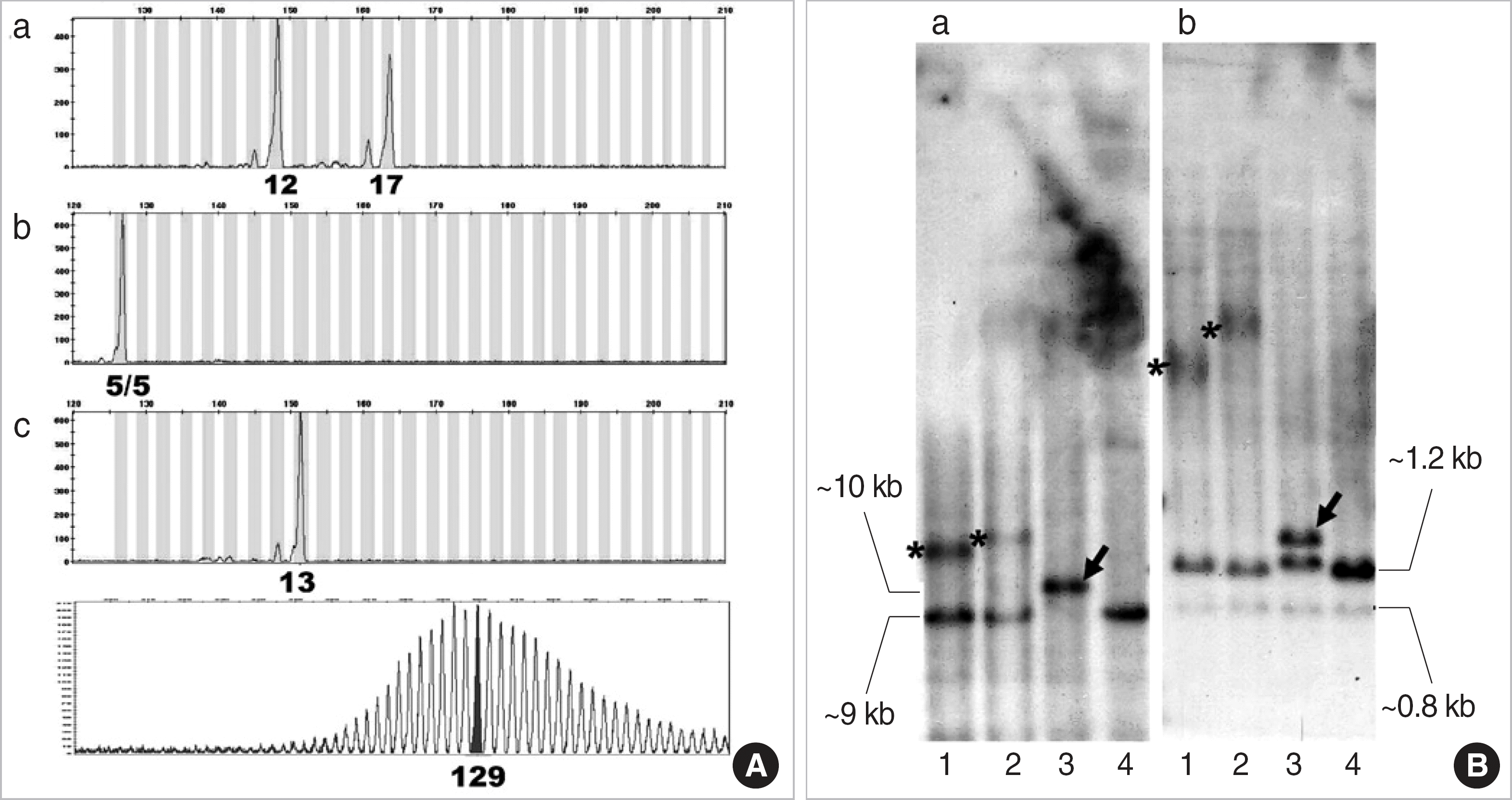
Fig. 3.
Pedigrees of 18 Korean DM1 families with two or more patients. Identifier number of each family is indicated above, and the CTG repeats number in the DMPK gene is below each symbol of family member.
Circle, female; square, male; diamond, offspring with no information about sex; filled symbol, symptomatic; open symbol, asymptomatic; shaded symbol, affected status not known; P, fetus in pregnant status.
Fig. 4.
The distribution of 430 normal alleles with different CTG repeats in the DMPK gene. Histogram is derived from the analysis of 94 normal alleles from 47 normal control subjects, 239 normal alleles from 120 unrelated subjects suspicious as DM1 but harbored no expanded alleles of 50 or more CTG repeats, and 97 normal alleles from 97 unrelated DM1 patients diagnosed by molecular analysis.
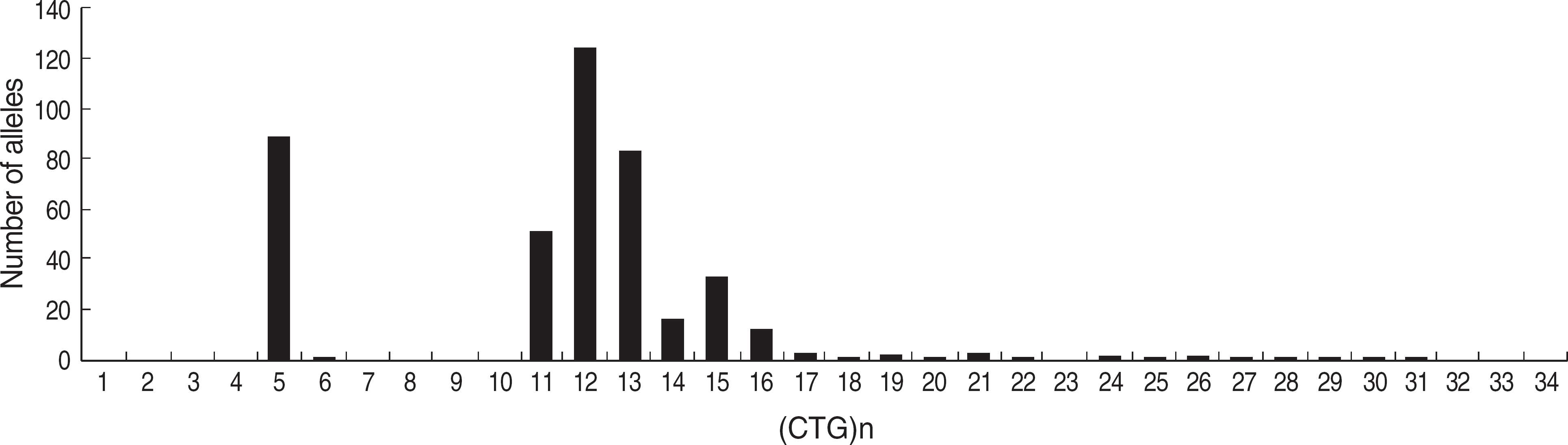
Fig. 5.
The CTG repeat expansion and the age of onset in 83 DM1 patients. Two factors show an inverse correlation.
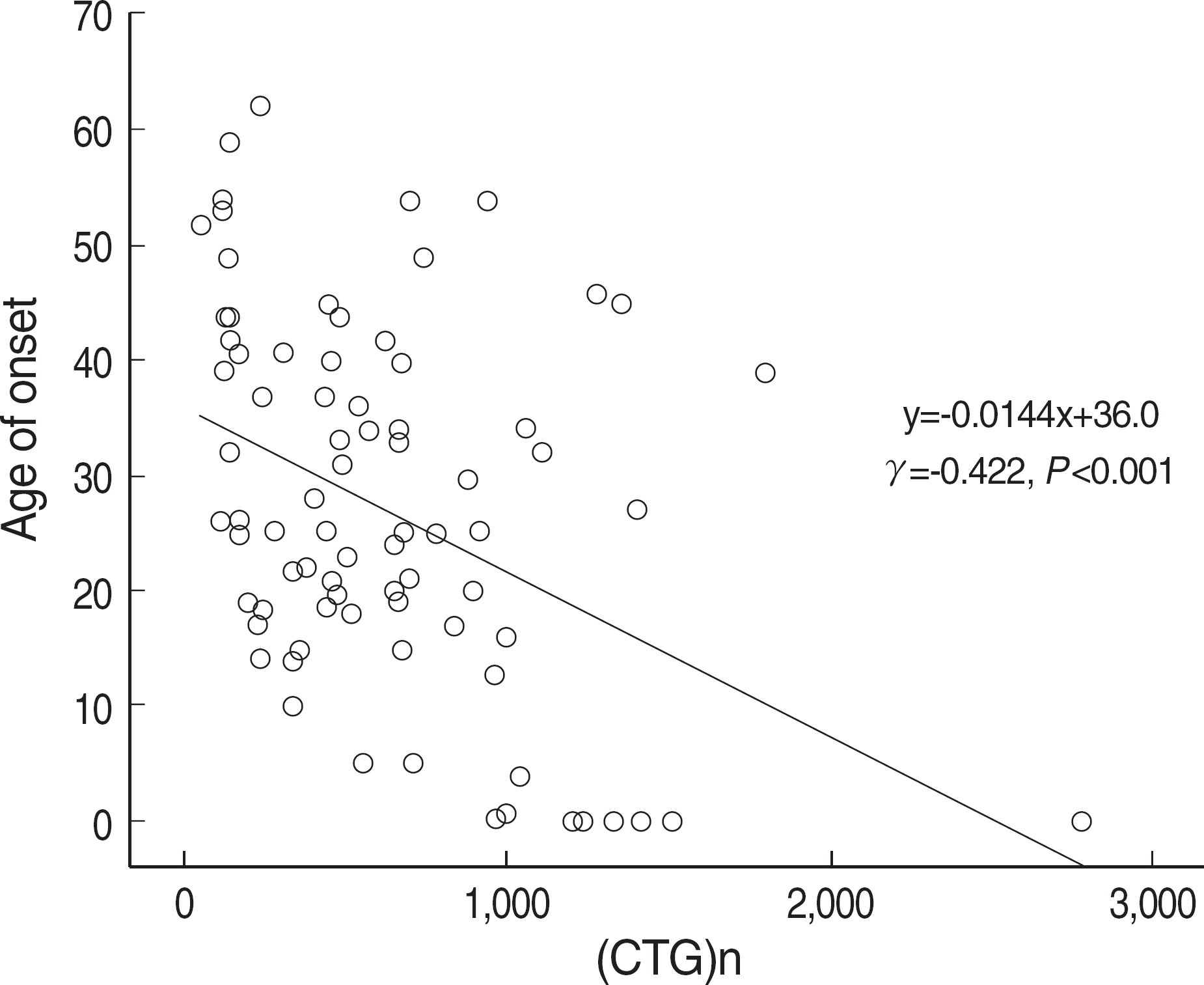
Fig. 6.
The size of CTG repeats expansion for (A) mother-offspring, and (B) father-offspring pairs. Anticipation was more evident in maternally-transmitted alleles. Contraction occurred only in paternally-transmitted alleles. Diagonal lines mean that the same allele size was expected between parent and offspring. Open circles located below the diagonal line indicate contraction of CTG repeats in offspring.
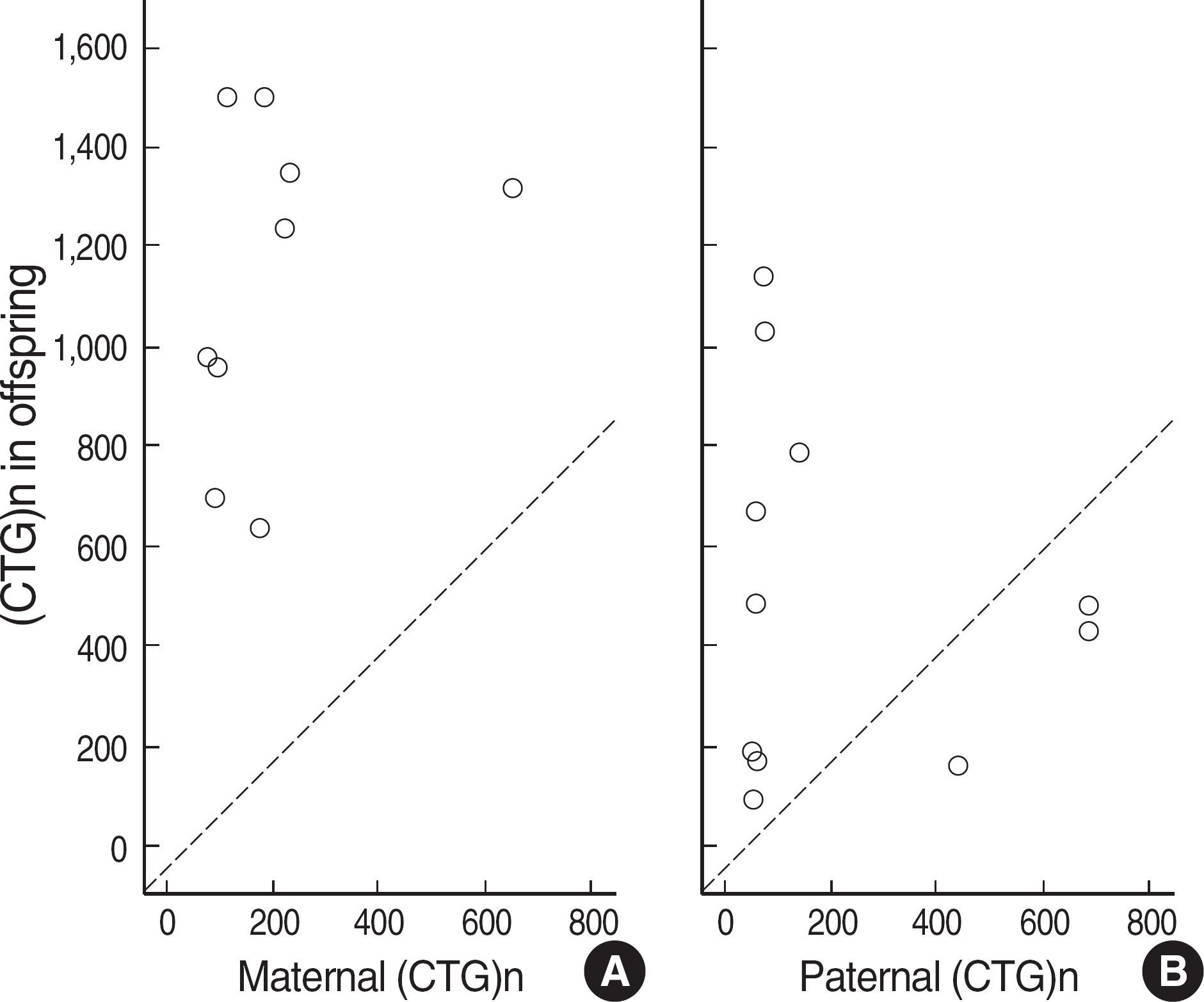
Table 1.
The distribution of CTG expansions in 125 Koreans
| Molecular diagnosis | Clinical phenotype∗ | CTG repeat | N |
|---|---|---|---|
| Premutation (mutable normal) | 35-49 | 1 | |
| DM1 | Mild | 50-99 | 9 |
| 100-149 | 13 | ||
| Classic | 150-999 | 82 | |
| 1,000-1,999 | 19 | ||
| Congenital | >2,000 | 1 | |
| Total DM1 patients | 124 |
Table 2.
Clinical characteristics in Korean DM1 patients confirmed by molecular tests
| Clinical findings | N∗ | Posivite | Negative | P | Comment | ||
|---|---|---|---|---|---|---|---|
| N (%) | CTG expansion median (range) | N | CTG expansion median (range) | ||||
| Myotonia | 105 | 84 (80.0) | 495 (50-2,770) | 21 | 750 (73-1,500) | >0.05 | |
| Muscular weakness | 105 | 81 (77.1) | 560 (50-2,770) | 24 | 280 (73-1,420) | 0.019 | |
| Family history | 88 | 46 (52.3) | 665 (118-2,770) | 42 | 470 (110-1,420) | >0.05 | |
| Muscular atrophy | 105 | 44 (41.9) | 540 (110-1,700) | 61 | 480 (50-2,770) | >0.05 | |
| Frontal baldness | 105 | 35 (33.3) | 550 (50-1,700) | 70 | 480 (73-2,770) | >0.05 | |
| CNS dysfunction/developmental delay | 35 | 17 (48.6) | 1,190 (230-2,770) | 18 | 410 (110-1,790) | 0.005 | |
| Cataract | 56 | 24 (42.9) | 638 (120-2,770) | 32 | 655 (110-1,400) | >0.05 | |
| Endocrine abnormalities | 44 | 17 (38.6) | 510 (120-990) | 27 | 450 (50-1,350) | >0.05 | |
| Cardiac abnormalities | 76 | 26 (34.2) | 590 (120-1,420) | 50 | 530 (110-2,770) | >0.05 | |
| Electromyography | 72 | 69 (95.8) | 510 (110-2,770) | 3 | 550 (230-1,240) | >0.05 | |
| Muscle biopsy | 23 | 19 (82.6) | 560 (140-2,770) | 4 | 295 (120-530) | >0.05 | |
| Creatine kinase | 50 | 17 (34.0) | 620 (140-1,700) | 33 | 550 (120-1,500) | >0.05 | Median 228.0 Range 68.0-1,044.0 (IU/L) |
Table 3.
The instability in CTG repeat expansion assessed by comparison between siblings in nine DM1 families
 XML Download
XML Download