

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
Wilson disease (WD) is one of the most common inborn errors of metabolism characterized by degenerative changes in the brain, liver and kidney dysfunction, and Kayser-Fleischer rings due to toxic accumulation of copper. We investigated a Korean family with WD occurred in two consecutive generations. The proband, a 14-yr-old girl, was noticed to have abnormal liver function on a routine health examination at school and was diagnosed of having WD by further laboratory tests and liver biopsy. Molecular genetic analysis of ATP7B gene demonstrated that she was homozygous for Ala-874Val mutation, one of the three common mutations in Korean patients with WD. Further study for her family members revealed that the proband's father, a paternal uncle, and the youngest sister were compound heterozygous for Ala874Val and Asn1270Ser mutations of the ATP7B gene. In addition, the proband's mother and a younger sister were heterozygous carriers of Ala874Val mutation. Therefore, WD occurred in two consecutive generations due to a WD father and a heterozygous mother. Actually, abnormal results on liver function tests were found in the proband's father and a paternal uncle a few years ago but a diagnosis of WD has not been made. Therefore, although WD has been thought to be uncommon in Korea, it should be considered in a differential diagnosis of patients exhibiting abnormal liver function with unknown cause.
Go to : 
References
1. Gitlin JD. Wilson disease. Gastroenterology. 2003; 125:1868–77.

2. Gaffney D, Fell GS, O'Reilly DS. ACP Best Practice No 163. Wilson's disease: acute and presymptomatic laboratory diagnosis and monitoring. J Clin Pathol. 2000; 53:807–12.
3. Kim EK, Yoo OJ, Song KY, Yoo HW, Choi SY, Cho SW, et al. Identification of three novel mutations and a high frequency of the Arg778Leu mutation in Korean patients with Wilson disease. Hum Mutat. 1998; 11:275–8.
4. Moon JS, Ko JS, Seo JK. Long-term clinical follow-up of Korean children with Wilson disease; twenty years' experience. J Korean Pediatr Soc. 2001; 44:127–38.
5. Yoo HW. Identification of novel mutations and the three most common mutations in the human ATP7B gene of Korean patients with Wilson disease. Genet Med. 2002; 4(S6):):S43–8.
6. Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993; 5:327–37.
7. Petrukhin K, Fischer SG, Pirastu M, Tanzi RE, Chernov I, Devoto M, et al. Mapping, cloning and genetic characterization of the region containing the Wilson disease gene. Nat Genet. 1993; 5:338–43.
8. Tanzi RE, Petrukhin K, Chernov I, Pellequer JL, Wasco W, Ross B, et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet. 1993; 5:344–50.
9. Chuang LM, Wu HP, Jang MH, Wang TR, Sue WC, Lin BJ, et al. High frequency of two mutations in codon 778 in exon 8 of the ATP7B gene in Taiwanese families with Wilson disease. J Med Genet. 1996; 33:521–3.
10. Kusuda Y, Hamaguchi K, Mori T, Shin R, Seike M, Sakata T. Novel mutations of the ATP7B gene in Japanese patients with Wilson disease. J Hum Genet. 2000; 45:86–91.
11. Shimizu N, Nakazono H, Takeshita Y, Ikeda C, Fujii H, Watanabe A, et al. Molecular analysis and diagnosis in Japanese patients with Wilson's disease. Pediatr Int. 1999; 41:409–13.
12. Firneisz G, Szonyi L, Ferenci P, Gorog D, Nemes B, Szalay F. Wilson disease in two consecutive generations: an exceptional family. Am J Gastroenterol. 2001; 96:2269–71.
13. Firneisz G, Szonyi L, Ferenci P, Willheim C, Horvath A, Folhoffer A, et al. The other mutation is found: follow-up of an exceptional family with Wilson disease. Am J Gastroenterol. 2004; 99:2504–5.
14. Yang TJ, Ji GH, Song MS, Hwang TG. The study of the initial presentation of Wilson disease at diagnosis. Korean J Pediatr Gastroenterol Nutr. 2001; 4:199–206.
15. Sokol RJ, Narkewicz MR. Copper and iron storage disorders. Suchy FJ, Sokol RJ, Balistreri WF, editors. Liver disease in children. 2nd ed.Philadelphia: Lippincott Williams & Wilkins;2001. p. 599–618.
16. Hefter H, Weiss P, Wesch H, Stremmel W, Feist D, Freund HJ. Late diagnosis of Wilson's disease in a case without onset of symptoms. Acta Neurol Scand. 1995; 91:302–5.
17. Wang XP. Wilson disease: asymptomatic or late-onset type? Acta Neurol Scand. 1996; 94:421–2.
18. Ferenci P. Regional distribution of mutations of the ATP7B gene in patients with Wilson disease: impact on genetic testing. Hum Genet. 2006; 120:151–9.
19. Gu YH, Kodama H, Du SL, Gu QJ, Sun HJ, Ushijima H. Mutation spectrum and polymorphisms in ATP7B identified on direct sequencing of all exons in Chinese Han and Hui ethnic patients with Wilson's disease. Clin Genet. 2003; 64:479–84.
20. Shimizu N, Kawase C, Nakazono H, Hemmi H, Shimatake H, Aoki T. A novel RNA splicing mutation in Japanese patients with Wilson disease. Biochem Biophys Res Commun. 1995; 217:16–20.
21. Nanji MS, Nguyen VT, Kawasoe JH, Inui K, Endo F, Nakajima T, et al. Haplotype and mutation analysis in Japanese patients with Wilson disease. Am J Hum Genet. 1997; 60:1423–9.
22. Ferenci P. Wilson disease. Indian J Gastroenterol. 2001; 20(S6):):S71–8.
23. Thomas GR, Forbes JR, Roberts EA, Walshe JM, Cox DW. The Wilson disease gene: spectrum of mutations and their consequences. Nat Genet. 1995; 9:210–7.
Go to :
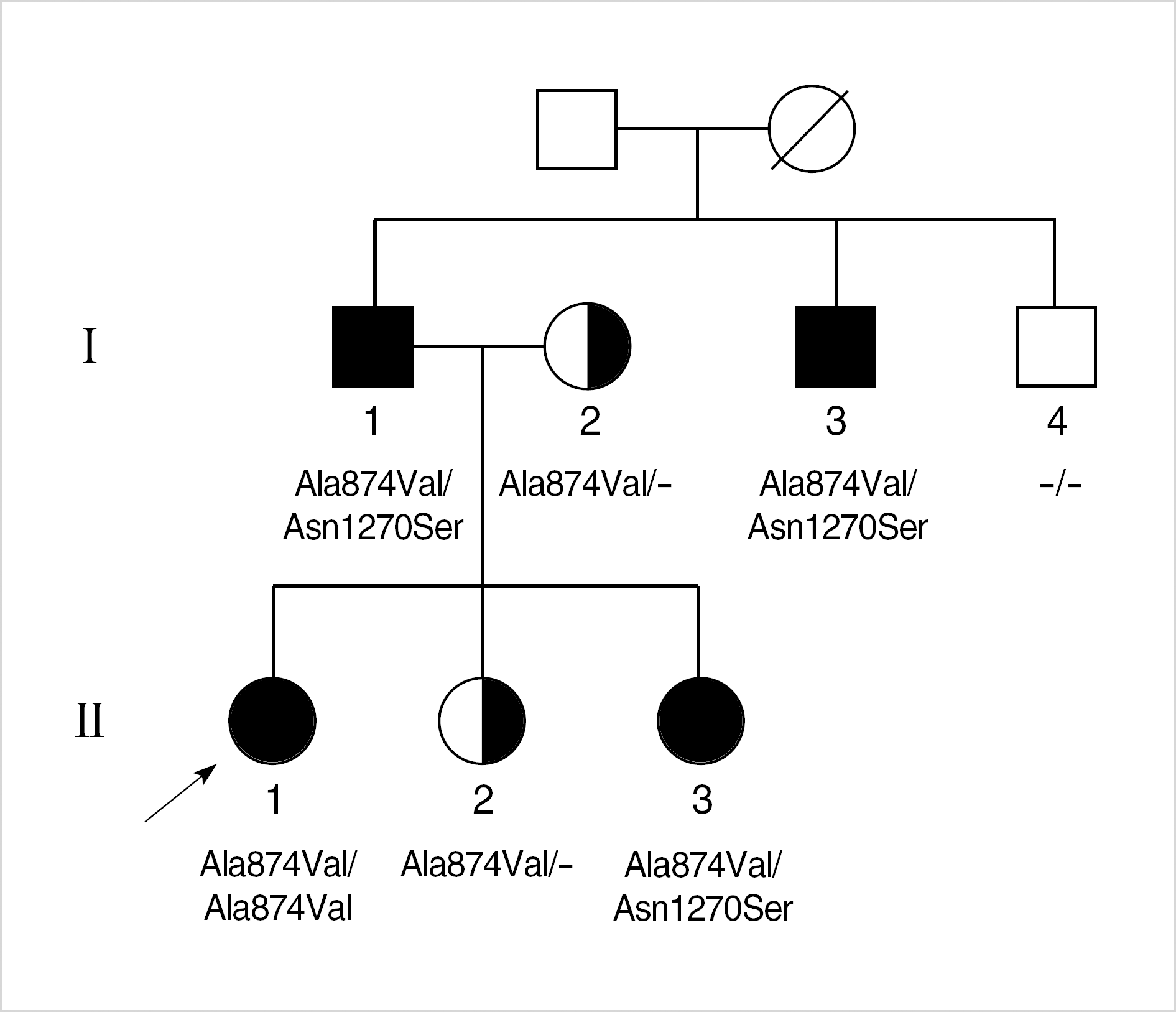 | Fig. 1.Pedigree of the family with Wilson disease. Mutations in the ATP7B gene identified in each family member are indicated. Circle, female; square, male; filled symbol, affected; half-filled symbol, heterozygous carrier; arrow, proband. |
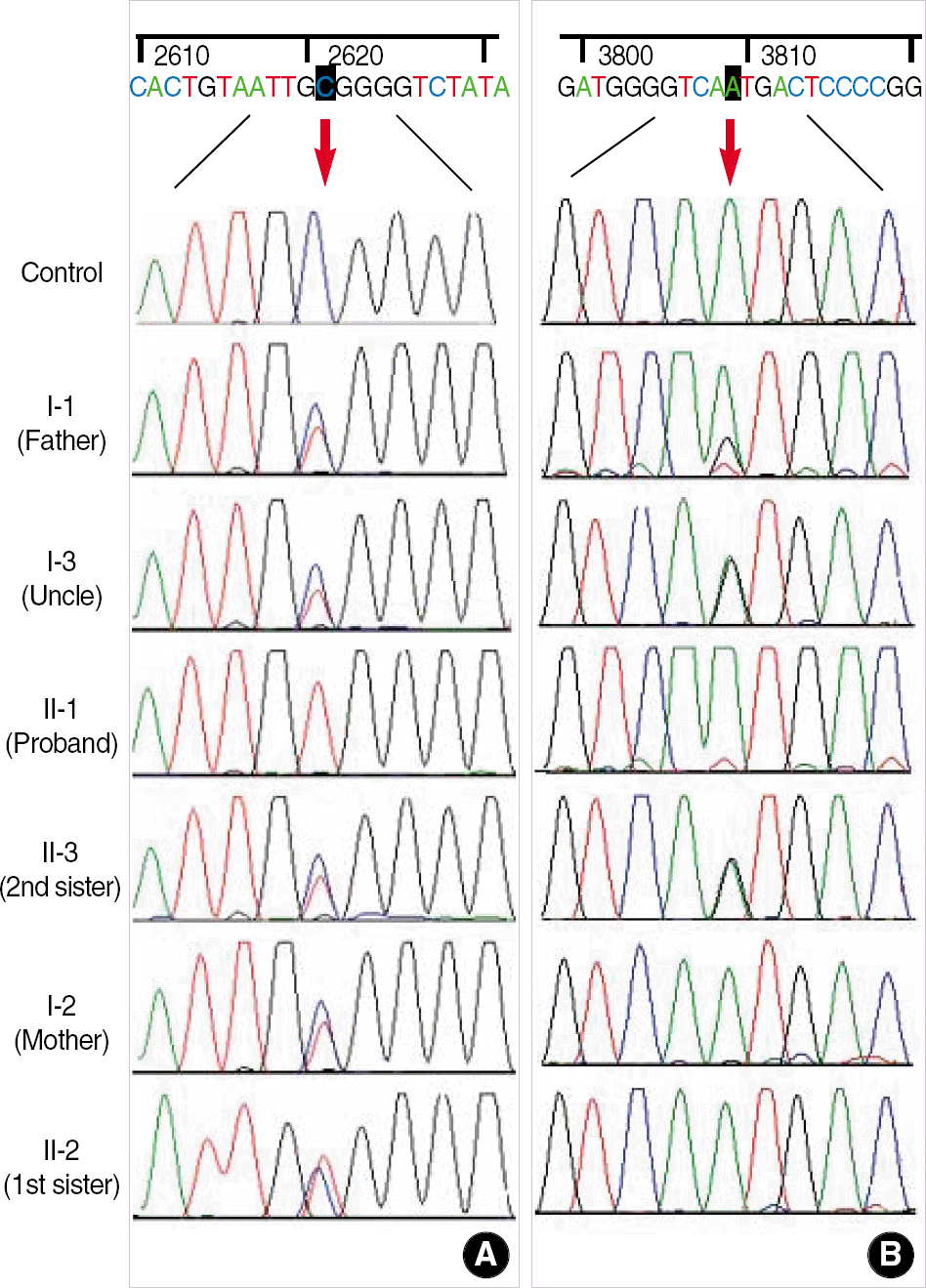 | Fig. 2.Result of gene analysis ATP7B. (A) Direct sequencing of exon 11. Arrow shows peak from the nucleotide 2621 position due to heterozygotic or homozygotic C to T transition. (B) Direct sequencing of exon 18. Arrow shows peak from the nucleotide 3809 position due to heterozygotic or homozygotic A to G transition. |
Table 1.
Clinical course of Wilson disease and laboratory features at diagnosis
 XML Download
XML Download