

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hepatic ischemia/reperfusion (I/R) injury is a pathophysiologic process whereby hypoxic organ damage is accentuated following the return of blood flow and oxygen delivery. Transient episodes of tissue ischemia are encountered during solid organ transplantation, trauma, hypovolemic shock and elective liver resection, when inflow occlusion or total vascular exclusion is used to minimize blood loss. The pathophysiology of hepatic I/R injury includes direct cellular damage as the result of ischemic insult as well as delayed dysfunction and damage that results from activation of inflammatory pathways.(1) Moreover, recent studies have suggested that hypoxia has been proposed as an inflammatogen such as lipopolysaccharide (LPS), as it can prime the innate immune system for an exaggerated inflammatory response through as yet unclear mechanisms.(2)
The Toll-like receptors (TLRs) play a critical role in early innate immunity toward invading pathogens and tissue damage by recognizing structural motifs that are only expressed on microbial pathogens called pathogen-associated molecular pattern (PAMP) or endogenous molecules released by damaged tissues (DAMP).(3) Previous studies suggested functional TLR4 signaling is required for High Mobility Group Box 1 (HMGB1) release from hepatocytes, which is an early mediator of injury and inflammation in hepatic warm I/R injury.(4,5) However, the mechanism for TLR4 signaling by hypoxia during initial hepatic I/R has not been elucidated.
The lipid rafts are a collection of lipid membrane microdomains characterized by insolubility in non-ionic detergent.(6) They range from a few nanometers to a few hundred nanometers in diameter and represent 50% of cellular membrane.(7) Lipid rafts serve as a platform where receptor-mediated signal transduction is initiated.(8) Recently, it has been postulated that membrane microdomains can be important in LPS signaling. Upon stimulation with LPS, the recruitment membrane-associated TLRs, such as TLR4 and TLR2, and other components of TLR complex occurs into the lipid rafts.(9)
In this study, we determined whether active TLR4 signaling is mediated by the receptor recruitment into lipid rafts following transient hypoxia in hepatic I/R injury and if these processes are required for the release of HMGB1, an early mediator of injury and inflammation in hepatic warm I/R injury.
METHODS
1) Materials
LPS (Escherichia coli 0111:B4) was purchased from List Biological Laboratories (Vandell Way, CA, USA). This LPS does not contain a significant amount of contaminating proteins that could stimulate TLR2 nonspecifically.(10) Williams Medium E was purchased from Gibco-BRL (Grand Island, NY, USA); fetal calf serum was purchased from HyClone Laboratories (Logan, UT, USA). The cholesterol sequestering agent, nystatin, obtained from Calbiocam (San Diego, CA, USA) was dissolved in sterile H2O at a concentration of 5 mg/ml. Filipin obtained from Cayman Chemical (Ann Arbor, MI, USA) was dissolved in dimethylsulfoxide (DMSO) at a concentration of 1 mg/ml. N-acetylcysteine (NAC) obtained from Sigma-Aldrich (St. Louis, MO, USA) was dissolved in sterile H2O at a concentration of 100 mM.
2) Animals
Male wild-type (C57BL/6) mice (8~12 week old) were purchased from The Jackson Laboratory. All animals were maintained in a laminar-flow, specific pathogen-free atmosphere at the University of Pittsburgh, PA, USA. Animal protocols were approved by the Animal Care and Use Committee of the University of Pittsburgh, PA, USA and the experiments were performed according to the National Institutes of Health guidelines for the use of laboratory animals.
3) Hepatocyte isolation
Hepatocytes were isolated from normal mice by an in situ collagenase (Type IV, Sigma-Aldrich) perfusion technique and modified as described previously.(11) Hepatocytes were separated from non-parenchymal cells by two cycles of differential centrifugation (50 g for 2 min) and further purified over a 30% Percoll gradient. Hepatocyte purity exceeded 98% as assessed by light microscopy, and viability typically was >95% as determined by tryptopan blue exclusion assay.
4) Cell culture and treatment
Hepatocytes (3×106) were plated onto 6-cm gelatin-coated plastic tissue culture dishes. The initial culture medium was Williams medium E containing 10% calf serum, 15 mM Hepes, 2 mM L-glutamine, and 100U/ml of penicillin and streptomycin. Hepatocytes were allowed to attach to plates overnight. For experiments involving hypoxia, the medium was replaced with hypoxic medium (equilibrated with 1% O2, 5% CO2, and 94% N2) and placed into a modular incubator chamber (Billups-Rothenberg), which was flushed with the same hypoxic gas mixture. The cells were exposed to hypoxia for various time points to demonstrate that TLR4 complex is translocated to the lipid rafts or HMGB1 release in the supernatants. For H2O2 studies, 3×106 cell were plated onto 6-cm gelatin-coated plastic tissue culture dishes and allowed to attach to the plates overnight. The media was replaced before stimulation of the cells with the appropriate amount of H2O2 and incubated at 37℃ for the times indicated. Where applicable, hepatocytes treated with LPS, NAC and nystatin or filipin were allowed to incubate for 30 min or 1 hour before hypoxic treatment.
5) Isolation of lipid raft using sucrose gradient centrifugation and detection of lipid raft by dot blot
Mouse Hepatocytes were harvested and scraped off in ice-cold PBS and lysed in 1 ml TKM buffer (50 mM Tris, pH 7.4, 25 mM KCl, 5 mM MgCl2, and 1 mM EDTA) containing 0.5% wt/vol Brij58 (Sigma-Aldrich, MO, USA) and protease inhibitors (Roche Diagnostics, IN, USA), incubated on ice for 30 min. 1 ml of 80% sucrose solution in TKM buffer was mixed with the lysate and this was overlaid with 5.5 ml of 35% sucrose solution in TKM buffer, followed by 2.5 ml of 5% sucrose solution in TKM buffer. After the samples were centrifuged at 38,000 rpm overnight in a Beckman SW41 rotor at 4℃, 1 ml fractions were collected from the top of gradient in each tube. An equal volume of each fraction was diluted in 100µl TBS-T buffer (20 mM Tris, pH 7.4, 150 mM NaCl; 0.05% Tween 20) and loaded onto Protran Nitrocellulose Sheet (0.45-µm pore; S&S BioScience, NH, USA) using Dot Blot (Bio-Rad Laboratories, CA, USA). The membrane was blocked in 5% milk in TBS-T, followed by incubation with CTxB-horseradish peroxidase (1:5,000; List Biological Laboratories, CA, USA). The third fraction from the top of gradient in the interface between 5% and 35% sucrose solutions designated the lipid raft layer.
6) Confocal immunofluorescence imaging
Hepatocytes on coverslips were fixed with 3% paraformaldehyde for 20 min, permeabilized with 0.1% Triton X-100 in PBS for 15 min, blocked with 2% BSA in PBS for 1 h, followed by three washes with 0.5% BSA in PBS. This was followed by anti-TLR4 antibody (1:200 in 0.5% BSA; Santa Cruz, CA, USA) for 60 min at room temperature. Secondary antibody (goat anti-rabit FITC- or Cy3-conjugated Fab fragment, 1:500 in 0.5% BSA; Invitrogen, CA, USA) was applied. For the purpose of demonstrating lipid raft layers, GM-1 ganglioside, a component of lipid rafts, was detected by incubating with rhodamine (TRITC)-conjugated CTxB (1:500; List Biological Laboratories, CA, USA). The slides were rinsed with PBS and coverslipped with gelvatol, a water-soluble mounting media (21 g polyvinylalcohol, 52 ml water, sodium azide, 106 ml 0.2-M Tris buffer). The slides were visualized with a confocal microscope (Fluoview 1000; Olympus).
7) Sodium dodecyl sulfate/polyacrylamide gel electrophoresis and Western blotting
Equal portions of each fraction were analysed by SDS/PAGE and transferred on to Immobilon P membranes (Millipore; Billerica, MA, USA) for 2 h at 250 mA in the presence of a transfer buffer. After transfer, the membrane was blocked for 1 h at room temperature with 5% milk in TBS-T buffer and incubated with a primary antibody to TLR4 (1:1,000; eBioscience, CA, USA), anti-MD2 (1:5,000; ProSci Inc., CA, USA), anti-CD14 (1:1,000; R&D System, MN, USA) and anti-CD11b/CD18 (1:250; Abcam, MA, USA) respectively for 12 h at 4℃. Blots were incubated at room temperature for 1 h with a horseradish peroxidase-conjugated secondary antibody against the primary antibody. After extensive washing with TBS-T buffer, membranes were developed with the Supersignal West Pico Chemiluminescent Kit (Pierce Chemical Co., IL, USA) and exposed to film.
For analysis of HMGB1 secretion, media from cells were centrifuged through Centricon (Millipore; Billerica, MA, USA) 10-kDa filter devices for 30 min at 14,000 g. Supernatants were collected from the top reservoir of the filter device and the process through the Centricon (Millipore; Billerica, MA, USA) 50-kDa filter devices was repeated for 15 min at 4,000 g. After protein concentration was determined using a BCA protein assay, equal volumes of elute were fractioned by SDS-PAGE similarly and transferred to nitrocellulose membranes. Following the initial blockade in 5% milk, the membranes were probed with polyclonal rabbit anti-HMGB1 (1:1,000; Abcam, MA, USA) antibody overnight at 4℃. Bound antibody was detected using a 1:5,000 dilution of goat anti-rabbit horseradish peroxidase-conjugated antibody (Pierce Chemical Co., IL, USA) and developed as described previously.
RESULTS
1) Hypoxia causes TLR4/MD2 complex translocation to lipid rafts associated with CD14
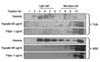
Recently, lipid rafts have been described as an important contributor to membrane associated recruitment and clustering of signal molecules.(12) In addition, lipid rafts play a role in LPS-induced signaling in macrophages.(13,14) Therefore, we evaluated whether hypoxia also causes LPS recognition receptors clustering within lipid rafts as with LPS stimulation. To substantiate TLR4 translocation to lipid rafts, we isolated raft fractions using discontinuous sucrose gradient ultracentrifugation of hepatocytes.(15) Effective isolation of membrane rafts was determined by the presence of GM1 ganglioside in the nonsoluble portion - fractions 3 to 5 of the sucrose gradient (Fig. 1A, top row). Hepatocytes were cultured under normoxic (21% oxygen) and hypoxic (1% oxygen) conditions and in the presence or absence of LPS stimulation. TLR4 was detected in detergent-soluble fractions, but not in lipid raft fractions, in normoxic cells (Fig. 1A, second row). After stimulation for 1 hour with LPS or for 1 hour and 6 hours with hypoxia, it was found that TLR4 translocates to lipid raft fractions. The translocation of TLR4 was time-dependent and increased under hypoxic conditions (Fig. 1A lower row). To further establish the clustering of the TLR4 receptor complex under hypoxia, we investigated if TLR4 adaptor protein, MD2, translocates to lipid rafts as shown in the case of TLR4. The MD2 also confined within the detergent-soluble fractions in normoxic cells translocates to lipid rafts after LPS or hypoxic stimulation (Fig. 1B). This observation suggests that adaptor protein MD2 associated with TLR4 clustering within lipid rafts is critical for TLR4 signaling in hypoxia as previously described in LPS response signaling.(16) In addition, we determined whether CD14 has a role in TLR4/MD2 association with lipid rafts. CD14 was found in lipid rafts regardless of normoxia and hypoxia (Fig. 1C). It could be postulated that CD14 association with lipid rafts may be a common element, where the CD14-lipid raft association provides a platform for recruitment of TLR4 complex.
These findings were verified using confocal immunofluorescence imaging. We stained hepatocytes under normoxic (21% oxygen) and hypoxic (1% oxygen) conditions with rhodamine-conjugated cholera toxin B (CTx B) and FITC - conjugated TLR4 after fixation and then permeabilized. The hepatocyte under the normoxic condition displayed diffuse intracellular TLR4 staining, whereas GM1, ganglioside, a classic raft marker, was mainly localized to the cell surface. In contrast, some TLR4 in hepatocytes after hypoxic stimulation displayed a peripheral staining and it colocalized with GM1 as shown by the overlay image (Fig. 1D).
2) Disruption of lipid raft layer blocks hypoxia induced TLR4/MD2 complex translocation
In order to investigate whether raft integrity affects TLR4 downstream signaling, we attempted to disrupt lipid rafts formation. The cells were then pre-incubated with either nystatin (60µg/ml), a fungal metabolite that binds membrane cholesterol and disrupts raft integrity or filipin (1µg/ml), a compound that sequestrates cholesterol in components of lipid rafts for 30 min or 1hour before exposure to hypoxia (1% oxygen).(17) It was found that nystation or filipin blocked TLR4/MD2 complex translocation to lipid rafts (Fig. 2). The raft disrupting drugs were found not only to displace the receptor complex involved in TLR4 and MD2 but also their signaling machinery, lending more support to the notion that lipid rafts are areas of concentrated receptor signaling.
3) Hypoxia causes β2-Integrin translocation to lipid rafts
As "adhesion molecules," the β2-Integrin (CD11b/CD18) acts as traditional receptors that promote cellular adhesion. Like most transmembrane receptors, β2-Integrin (CD11b/CD18) is capable of transmitting signals elicited by ligand binding.(18) Recent reports indicate that three membrane-associated proteins, CD14, β2-Integrin (CD11b/CD18), and Toll-like receptor (TLR) 4, may serve as LPS recognition and/or signaling receptors in murine macrophages.(19) We investigated whether β2-Integrin (CD11b/CD18) as transmembrane receptor associates with TLR4 complex in lipid rafts of hepatocytes under the hypoxic stimulation. It was found that β2-Integrin (CD11b/CD18) also translocated to lipid rafts under hypoxic stimulation (Fig. 3). These findings suggest that for expression of a full repertoire of hypoxic induced TLR4 signaling, TLR4/MD2 complex and β2-Integrin (CD11b/CD18) must be coordinately engaged to deliver optimal signaling by translocation to lipid rafts of hepatocyes.
4) The TLR4/MD2 complex translocation is ROS dependent signaling
We next asked whether exposure of hepatocyte to oxidants could mimic the effect of hypoxia on TLR4 signaling. Hepatocytes were treated with H2O2 in concentration from 50~500µM. The H2O2 was shown to induce a dose and time-dependent, increased TLR4/MD2 translocation to lipid rafts, with slight effect at 50µM for 1-hour exposure (not depicted) and marked increased translocation at 100~500µM up to 6 hours (Fig. 4A). To confirm the role of ROS in TLR4 signaling, we added N-acethylcystein (NAC) to the cell culture medium before hypoxic stimulation. NAC blocked both TLR4 and MD2 translocation to lipid rafts (Fig. 4B). The inhibition of TLR4/MD2 translocation to lipid rafts by NAC determined a role for oxidative stress in the effect.
5) Disruption of lipid raft layer blocks hypoxia induced HMGB1 release
In order that we determine the receptor clustering within the lipid rafts, which is required for TLR4/HMGB1 signaling in initial hepatic warm I/R injury, we conclusively asked whether hypoxia induced HMGB1 release could be blocked by disrupting lipid rafts. The cells were then pre-incubated with nystatin (60µg/ml) for 30 min or 1 hour. Hepatocytes were cultured under hypoxic (1% oxygen) conditions for 24 hours. Recently, previous published studies state that the release of HMGB1 is increased up to 24 hours hypoxia by active regulated process without definite cell necrosis.(4) We attempted to analyze HMGB1 after concentrating media. It was found that the release of HMGB1 is blocked by nystatin as shown by NAC, a scavenger of ROS (Fig. 5A). Contrary to the amount of HMGB1 release in media, it was also determined that more quantities of intracellular HMGB1 in cells of disrupting lipid rafts is present than in hypoxic cell without disrupting lipid raft (Fig. 5B). These results suggest that the ability of components of lipid raft to modify HMGB1 releasing mechanisms is involved in hepatic I/R injury.
DISCUSSION
More recent studies on hepatic I/R have focused on the link between innate immunity and the proximal events involved cell damage.(20) Several lines of evidence indicate that DAMPs (damage-associated molecular pattern) appear to accentuate innate immunity by engaging certain cell surface receptors such as TLR4.(21,22) The DAMPs-induced overwhelming immune activation can lead to exaggerated local and systemic inflammation that may extend tissue damage. In this study, we focused on the mechanism by which hypoxia, initial biologic signal in hepatic I/R injury could lead to activate TLR4 signaling in hepatocytes. These experiments demonstrate that hypoxia, through the generation of ROS, causes TLR4 complex translocation to the lipid rafts and that through the formation of these TLR4 complexes within lipid rafts, ultimately TLR4 downstream signaling is involved in the release of HMGB1, an early mediator of injury and inflammation in hepatic I/R injury.
The importance of lipid rafts in LPS induced TLR4 signaling in immune cell has also been indicated by the findings of LPS down-regulation of caveolin-1 and recruitment of signaling molecules to lipid rafts in MonoMac-6 cells upon LPS stimulation.(14) TLR4 signaling in epithelial cells differs in some ways from that in immune cell but a recent study showed that LPS-induced activation requires lipid rafts in epithelial cells.(9) Our data show that TLR4 and MD2 become translocated to lipid rafts under hypoxic stimulation, indicating that lipid rafts also play a critical role in hypoxia induced TLR4 signaling in hepatocytes. Interestingly, another LPS binding molecule, CD14, was always found in lipid rafts, regardless of hypoxia or normoxia. This finding indicates that CD14 is a GPI-anchored protein, most of which resides in lipid rafts, dynamic microdomains that are enriched in cholesterol and glycosphingolipids. Of interest, it remains to be determined whether CD14 has a role in TLR4 association with lipid rafts. The mechanisms by which CD14 amplifies TLR4 signaling remains to be explored. Some reports indicate that the recruitment of TLR4/MD2 to lipid rafts needs to precede LPS transfer from CD14 to TLR4/MD2 in LPS signaling.(23) However, multiple lines of evidence suggest that CD14 association with lipid rafts may be a common element where the CD14-lipid raft association provides a platform for recruitment of not only TLRs but also another key signaling molecules for potent interaction and signal initiation.(24)
A recent study by Perera et al. demonstrated another transmembrane receptor, β2-Integrin (CD11b/CD18), was also required for optimal induction of COX-2, IL-12 p35, and IL-12 p40 genes by low concentrations of LPS or by all concentrations of Taxol.(21) The data in this report indicate that for expression of a full repertoire of LPS/Taxol inducible genes, CD14, TLR4, and β2-Integrin (CD11b/CD18) must be coordinately engaged to deliver optimal signaling to the macrophage. This observation is also consistent with our finding that β2-Integrin (CD11b/CD18) translocates to lipid rafts under hypoxic stimulation. This result implies that interaction of all three molecules (CD14, TLR4/MD2, and CD11b/CD18) under the hypoxic stimulation is proposed to deliver a coordinated set of intracellular signals that results in the induction of the complete panel of hypoxia-inducible inflammatory genes.
How do hepatocytes sense hypoxic conditions? What is the hypoxia related DAMPs? In these studies, we used biochemical approaches to demonstrate that oxidative stress by H2O2 in vitro was able to induce TLR4/MD2 complex translocation to lipid rafts. Further confirmation of oxidative stress induced TLR4 signaling was provided in that N-acethylcystein (NAC) blocked both TLR4 and MD2 translocation to lipid rafts under hypoxic conditions. Our data indicate that hypoxia, through the generation of ROS is involved in this hypoxia-induced TLR4/MD2 translocation to lipid rafts. However, the source of the hypoxia-induced ROS production is unknown. Several reports showed that hypoxia increases ROS generation from mitochondria and these oxidant signals appear to act as second messengers in adaptive responses to hypoxia.(25,26) However, recent studies have suggested that NAD(P)H oxidase is an important source of ROS mediating organ injury after hemorrhagic shock.(27) These suggestions are consistent with those of Fan et al. who reported that tissue ischemia and reperfusion activates NAD(P)H oxidase in PMN through HMGB1/TLR4 signaling.(22) Based on experiments in other cells, further studies are necessary to determine the mechanism by which hypoxia, initial biologic signal in hepatic I/R injury, induces ROS production in hepatocytes.
HMGB1, a 30 kDa nuclear and cytosolic protein widely studied as a transcription factor and growth factor, has recently been identified as a cytokine mediator of lethal systemic inflammation (e.g. endotoxemia and sepsis), arthritis and local inflammation.(28) Under inflammatory or injurious conditions, HMGB1 can be released actively by innate immune cells (macrophages, monocytes), and passively by necrotic cells.(29) Initial experiments in hepatic I/R with HMGB1 elucidated hypoxic hepatocytes showed increased levels of HMGB1 expression and that inhibiting HMGB1 with a neutralizing antibody decreased hepatic damage following I/R(21). Furthermore, HMGB1 initiates inflammation in hepatic I/R through activation of TLR4 dependent signaling.(4) Therefore, It is possible that down regulation of TLR4 signaling leads to decreased releasing of HMGB1 from hepatocytes. In the present experiments, we observed decreased levels of HMGB1 from hepatocytes after 24 h hypoxia by inhibiting TLR4 complex translocation to lipid raft as well as antioxidant treatment. However, it is not clear from our studies whether HMGB1 is directly regulated by TLR4 signaling because there is a discrepancy of ischemic duration for optimal releasing of HMGB1 between in vivo and in vitro study. We previously reported that HMGB1 levels are increased during hepatic I/R as early as 1 h after reperfusion.(21) Further work is required to elucidate the mechanisms that are involved in TLR4/HMGB1 signaling.
In summary, these present experiments provide evidence that hypoxia induced ROS may exert its effects by regulating the distribution of the most proximal component of the signaling cascade, namely the TLR4 complex. Future studies defining the mechanism of oxidant-induced TLR4 redistribution may provide fundamental insights into the regulation of TLR4 in hepatic I/R models and suggest novel therapeutic approaches in disease states wherein the process of ischemia/reperfusion contributes to the pathogenesis of disease.




 XML Download
XML Download