

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Blood vessel walls are composed of endothelial cells, smooth muscle cells, and extracellular matrix. These components each play a role in maintaining vascular homeostasis. For example, vascular endothelial cells play a crucial role in maintaining tissue homeostasis and forming the interface between the blood and surrounding tissue.(1) However, dysfunction of these components can cause several vascular diseases. Abnormal proliferation of vascular smooth muscle cells and the resultant accumulation of extracellular matrix components, such as collagen, glycosaminoglycan, fibronectin, elastin, and laminin, in the arterial intima are pathological hallmarks of atherosclerotic vascular disease.(2)
Many studies have suggested that the extracellular matrix is involved in various cardiovascular pathologies. Extracellular matrix affects the activation, proliferation, death, and numerous activities of cardiac and vascular cells, resulting in cardiovascular system remodeling.(3) Collagens are extracellular matrix proteins that play an important role in maintaining the structural integrity of various tissues. There are at least 25 different types of collagens, and 13 types are known to be present in the blood vessel wall.(4)
Endothelial cells respond to various stimuli by releasing endothelium-dependent vasodilators.(5) Nitric oxide (NO) is an endothelium-dependent vasodilator, and NO is synthesized from arginine by nitric oxide synthase (NOS). NO produced by endothelial cells spreads to the subendothelial space and causes vascular smooth muscle cells to relax. NO activates soluble guanylyl cyclase (sGC) and increases the production of cyclic guanosine monophosphate (cGMP).(6) cGMP, the main second messenger in the L-arginine-NO pathway, mediates the biological activities of NO, such as vasodilatation, inhibition of platelet aggregation, leukocyte recruitment, and cell proliferation. NO activates further downstream cascades, such as cGMP-regulated protein kinase, ion channels, and phosphodiesterase (PDE).(7) Certain diseases or specific types of pharmacological stimulation can markedly alter NO-cGMP signaling.(8)
Cyclosporine A (CsA) is a potent immunosuppressive agent, and it has been used to prevent the rejection of transplanted organs and to treat autoimmune diseases. Many side effects of CsA, such as hypertension, nephrotoxicity, and hepatotoxicity, have been reported.(9) Coronary endothelial dysfunction, microvascular dysfunction, and deformation of microvasculature are known to be associated with CsA.(10) Ramzy et al.(11) reported that CsA impairs vascular homeostasis and causes transplant vasculopathy through induction of endothelial injury.
Pentoxifylline (PTX) is a non-selective PDE inhibitor, and it inhibits type 1 PDE to type 5 PDE. PTX has been used clinically in the treatment of peripheral vascular diseases due to its beneficial effects on blood rheology and microcirculation.(12)
In the present study, we investigated the effect of CsA on collagen synthesis in arterial endothelial cells and clarified whether PTX has a protective effect against CsA-induced arterial vasculopathy using calf pulmonary artery endothelial (CPAE) cells. This study was conducted using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, reverse transcription-polymerase chain reaction (RT-PCR), Western blot analysis, NO detection, and cGMP enzyme immunoassay.
METHODS
1) Drugs and reagents
CsA and PTX were purchased from Sigma Chemical Co. (St. Louis, MO, USA). The MTT assay kit was purchased from Boehringer Mannheim GmbH (Mannheim, Germany). A cGMP immunoassay kit was purchased from Sapphire Bioscience (Redfern New South Wales, Australia).
2) Cell culture
Cells derived from calf pulmonary arterial endothelium (CPAE) were purchased from the Korean Cell Line Bank (KCLB; Seoul, Korea). The cells were cultured in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco BRL, Grand Island, NY, USA) at 37℃ in 5% CO2, 95% O2 in a humidified cell incubator. The medium was changed every 2 days. The cells were plated onto culture dishes at a density of 2×104 cells/cm2 at 12 h prior to the PTX and CsA treatment.
3) MTT cytotoxicity assay
MTT cytotoxicity assay was performed in order to determine the cytotoxicity of CsA and PTX. CPAE cells were grown in a final volume of 100 µl culture medium per well in a 96-well plate. For the cytotoxicity of CsA, the cells were treated with CsA at concentrations of 0.001 µM, 0.01 µM, 0.1 µM, 1 µM, and 10 µM for 24 h. To investigate the cytotoxicity of PTX, the cells were treated with PTX at concentrations of 5 µM, 10 µM, 50 µM, 100 µM, 500 µM, and 1,000 µM. The cells in the control group were left untreated.
After 10 µl of the MTT labeling reagent containing 5 mg/ml MTT in phosphate-buffered saline (PBS) was added to each well, the plates were incubated for 4 h. Solubilization solution (100 µl) containing 10% sodium dodecyl sulfate in 0.01 M hydrochloric acid was added to each well, and the cells were incubated for another 12 h. The absorbance was then measured with a microtiter plate reader (Bio-Tek, Winooski, VT, USA) at a test wavelength of 595 nm with a reference wavelength of 690 nm. The optical density (O.D.) was calculated as the difference between the absorbance at the reference wavelength and that observed at the test wavelength. Percent viability was calculated as (O.D. of drug-treated sample/control O.D.) ×100.
4) RNA isolation and RT-PCR
RT-PCR was performed in order to identify the expression of collagen type I mRNA. Total RNA was isolated from CPAE cells using RNAzol™B (TEL-TEST, Friendswood, TX, USA). Two µg of RNA and 2 µl of random hexamers (Promega, Madison, WI, USA) were added together, and the mixture was heated at 65℃ for 15 min. One µl of AMV reverse transcriptase (Promega), 5 µl of 2.5 mM dNTP (Promega), 1 µl of RNasin (Promega), and 8 µl of 5×AMV RT buffer (Promega) were then added to the mixture, and the final volume was brought up to 40 µl volume with diethylpyrocarbonate (DEPC)-treated water. The reaction mixture was then incubated at 42℃ for 2 h.
PCR amplification was performed in a reaction volume of 40 µl containing 1 µl of the appropriate cDNA, 0.5 µl of each primer set at a concentration of 10 pM, 4 µl of 10×RT buffer, 1 µl of 2.5 mM dNTP, and 0.2 µl of Taq DNA polymerase (Takara, Shiga, Japan). For mouse collagen type I, the primer sequences were 5'-GAAAGGAGAGAGCGGCAAC-3' (a 19-mer sense oligonucleotide) and 5'-TCAATACCAGGGAGACCCAC-3' (a 20-mer anti-sense oligonucleotide). For GAPDH, the internal control used in the study, the primer sequences were 5'-TCTTCCAGGAGCGAGAT-3' (a 17-mer sense oligonucleotide) and 5'-ACAGACACGTTGGGAG-3' (a 16-mer anti-sense oligonucleotide). The expected size of the PCR product was 373 bp for collagen type I and 499 bp for GAPDH.
For collagen type I, the PCR procedures were carried out using a PTC-0150 MiniCycler (Bio-Rad, Hercules, CA, USA) under the following conditions: initial denaturation at 94℃ for 5 min, followed by 30 amplification cycles, each consisting of denaturation at 94℃ for 30 sec, annealing at 58℃ for 30 sec, and extension at 72℃ for 30 sec, with an additional extension step at 72℃ for 10 min at the end of the procedure. For GAPDH, PCR was carried out under the same conditions, except for the amplification cycles, as GAPDH was amplified with 25 cycles. The final amount of RT-PCR product for each of the mRNA species was calculated densitometrically using Molecular Analyst™ version 1.4.1 (Bio-Rad).
5) Western blot analysis
The cells were pre-treated with PTX for 1 h, followed by treatment with CsA. The cells were then collected and lysed in a lysis buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.5% deoxycholic acid, 1% Nonidet P40, 0.1% SDS, 1 mM PMSF, and 100 mg/ml leupeptin. Protein content was measured using a Bio-Rad colorimetric protein assay kit (Bio-Rad). A total of 30 µg of protein was separated on SDS-polyacrylamide gels and transferred onto a nitrocellulose membrane. Mouse collagen type I antibody (1 : 250; Santa Cruz Biotech, Santa Cruz, CA, USA) was used as a primary antibody. Horseradish peroxidase-conjugated anti-mouse antibody for collagen type I (1 : 2,000; Amersham Pharmacia Biotech GmbH, Freiburg, Germany) was used as a secondary antibody. Band detection was performed using the enhanced chemiluminescence (ECL) detection system (Amersham Pharmacia Biotech GmbH).
6) Determination of NO production
In order to determine the effect of CsA and PTX on NO production, the amount of nitrite, which is a by-product of nitric oxide transformation, in the supernatant was measured based on the Griess reaction as an indicator of NO production. The cells were pre-treated with PTX for 1 h and then treated with CsA and incubated for 24 h. After collection of 100 µl of cell culture medium, 50 µl of 1% sulfanilamide was added to each well, and the plate was incubated at room temperature for 10 min. 0.1% naphtylethylenediamine containing 5% phosphoric acid was then added, and the plate was incubated at room temperature for another 10 min. The absorbance of the content of each well was measured at a wavelength of 540 nm. The nitrite concentration was calculated from a nitrite standard curve generated by mixing 0 to 250 µM sodium nitrite solutions with Griess reagent. The standard curve was usually linear between 0 and 250 µM of sodium nitrite.
RESULTS
1) Effects of CsA and PTX on the viability of CPAE cells
CPAE cells were cultured with CsA and PTX at various concentrations in order to assess the cytotoxic effects of CsA and PTX in CPAE cells, and an MTT assay was then performed.
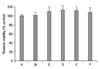
As shown in Fig. 1, the viabilities of the cells incubated with CsA at concentrations of 0.001 µM, 0.01 µM, 0.1 µM, 1 µM, and 10 µM for 24 h were 101.65±1.92%, 109.46±2.12%, 112.97±1.95%, 112.15±1.42%, and 107.52±3.94% of the control value, respectively. The viabilities of the cells treated with CsA at concentrations of 0.01 µM, 0.1 µM, and 1 µM were significantly increased in comparison to the control cells. The concentration of CsA was set at 1 µM for the next experiments.
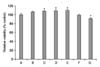
The viabilities of cells incubated with PTX at concentrations of 5 µM, 10 µM, 50 µM, 100 µM, 500 µM, and 1,000 µM for 24 h were 106.41±1.66%, 108.52±1.94%, 108.66±3.14%, 110.16±1.99%, 99.04±2.03%, and 91.67±1.65% of the control value, respectively (Fig. 2). The viabilities of the cells treated with PTX at concentrations of 10 µM, 50 µM, and 100 µM were significantly increased, while treatment with 1,000 µM PTX decreased the viability of the cells. The concentrations of PTX were set at 100 µM and 1,000 µM for the next experiments.
2) Effects of CsA and PTX on the mRNA expression of collagen type I
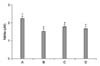
RT-PCR analysis of the mRNA level of collagen type I was performed in order to estimate of relative expression level of this gene. In the present study, the level of collagen type I mRNA in the control cells was set at 1.00. The level of collagen type I mRNA following treatment with 1 µM CsA for 24 h was 1.74±0.10. The levels of collagen type I mRNA in the cells pre-treated with PTX at concentrations of 100 µM and 1,000 µM for 1 h and then treated with CsA for 24 h were 0.94±0.05 and 1.24±0.06, respectively (Fig. 3).
In the present results, CsA treatment significantly increased the expression of collagen type I mRNA in the CPAE cells, while PTX pre-treatment suppressed the expression of collagen type I mRNA. Especially, 100 µM PTX decreased the expression of collagen type I mRNA to near the control level.
3) Effects of CsA and PTX on the protein expression of collagen type I
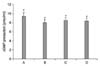
In order to estimate the relative expression of collagen type I protein by CsA and PTX, the level of collagen type I protein in the control cells was set at 1.00. The level of collagen type I protein after treatment with 1 µM CsA for 24 h was 3.45±0.15. The levels of collagen type I protein in cells pre-treated with PTX at concentrations of 100 µM and 1,000 µM for 1 h and then treated with CsA for 24 h were 1.13±0.10 and 1.81±0.09, respectively (Fig. 4).
In the present results, CsA treatment significantly increased the expression of collagen type I protein in the CPAE cells, while PTX pre-treatment suppressed the expression of collagen type I protein. Especially, 100 µM PTX decreased the expression of collagen type I protein to near the control level.
4) Effects of CsA and PTX on the production of NO
From the NO detection assay, the amount of nitrite was significantly decreased from the control level of 2.24±0.12 µM to 1.53±0.14 µM by treatment with 1 µM CsA for 24 h. However, the amounts of nitrite were slightly increased to 1.78±0.13 µM and 1.67±0.13 µM by pre-treatment with PTX at concentrations of 100 µM and 1,000 µM followed by treatment with CsA for 24 h (Fig. 5).
In the present results, CsA treatment significantly suppressed NO production in the CPAE cells. Pre-treatment with PTX slightly increased the production of NO; however, this increase was not statistically significant.
5) Effects of CsA and PTX on the production of cGMP
The results of the cGMP immunoassay showed that the amount of cGMP was significantly decreased from the control level of 9.34±0.39 pmol/ml to 7.97±0.28 pmol/ml by treatment with 1 µM CsA for 24 h. However, the amounts of cGMP were slightly increased to 8.44±0.31 pmol/ml and 8.34±0.29 pmol/ml by pre-treatment with PTX at concentrations of 100 µM and 1,000 µM for 1 h followed by treatment with CsA for 24 h (Fig. 6).
In the present results, CsA treatment significantly suppressed cGMP production in the CPAE cells. Pre-treatment with PTX slightly increased cGMP production, but this increase was not statistically significant.
DISCUSSION
The introduction of CsA ushered in a new era in the field of organ transplantation around the world. CsA treatment substantially improved the overall success rates after transplantation of solid organs, such as the kidney, heart, liver, and bone marrow. CsA has diminished the rejection rate after organ transplantation, and it is known to be effective in the treatment of autoimmune diseases.(13) However, hypertension, nephrotoxicity, hepatotoxicity, and various endothelial dysfunctions have been reported as side effects of CsA.(9,10) Impairment of vascular homeostasis and transplantation vasculopathy are also induced by CsA treatment.(11)
Although the underlying mechanisms of CsA-induced endothelial dysfunction and transplantation vasculopathy have not been fully elucidated, CsA is known to increase the expression of collagen type I in endothelial cells and to induce deposition of collagen type I.(14) Collagens are responsible for the tensile strength and elasticity of the blood vessel wall. In particular, type I and III collagens are most predominant in vessels, and collagen type I is known to impart arterial stiffness.(4) Excessive collagen accumulation is associated with vascular diseases and vascular injury.(15) Progressive accumulation of collagen content in the vascular wall is induced by an increase in collagen synthesis or decrease in collagen degradation. The results of the present study showed that CsA treatment significantly increased the levels of collagen type I mRNA and protein in CPAE cells.
Dooley et al.(16) suggested that abnormal NO regulation contributed to endothelial dysfunction in systemic sclerosis characterized by excessive deposition of collagens, and Dooley et al.(17) demonstrated that NO attenuated the secretion of collagen type I in human dermal fibroblasts. NO has also been shown to down-regulate the expression of extracellular matrix proteins, such as collagen type I, in dermal and cardiac fibroblasts, smooth muscle cells, and other types of cells.(18)
Many studies demonstrated that CsA impairs vasodilation and causes vasoconstriction.(19) CsA reduces the release of NO and cGMP, resulting in vasoconstriction.(20) It was suggested that CsA may alter the transcription of various genes, such as NOS, transforming growth factor-beta (TGF-β), endothelin-1, collagen I and IV, and Bcl-2.(21) In several studies, CsA treatment decreased endothelial NOS mRNA and protein expression.(11,22) CsA inhibits the transcription of inducible NOS in the vessels and impairs the production of NO and cGMP.(23) In the present study, CsA treatment inhibited NO synthesis and cGMP production in the CPAE cells. These results suggest that the CsA-induced reduction of NO and cGMP may have contributed to the increase in collagen type I in the CPAE cells.
PTX is a nonselective PDE inhibitor, and it has been shown to suppress a variety of cellular processes implicated in vascular lesion formation, such as platelet aggregation, vascular smooth muscle cell proliferation, apoptosis, nuclear factor-kappa B (NF-κB) activation, tumor necrosis factor-α (TNF-α) synthesis, and fibroblast collagen production. The actions of PTX occur by increasing the intracellular levels of cyclic adenosine monophosphate (cAMP) and cGMP.(24) Moreover, PTX suppresses the production of collagen type I by down-regulating the levels of transcription factors involved in collagen synthesis in vascular smooth muscle cells and hepatic stellate cells.(25) In the present study, PTX pre-treatment significantly decreased the levels of collagen type I mRNA and protein in CPAE cells. These results demonstrated that PTX is effective in the reduction of CsA-induced collagen type I synthesis.
PTX has been used to treat peripheral vascular diseases,(12) and PTX is known to be remarkably effective in situations where high blood pressure dominates the progression of renal diseases. The precise mechanisms of action by which PTX ameliorates vascular diseases remains elusive; however, PTX has been reported to increase the levels of intracellular cAMP, decrease blood viscosity, and dilate blood vessels. Berkenboom et al.(26) suggested that high concentrations of PTX may directly induce endothelium-derived NO-mediated vascular relaxation. Wang et al.(27) reported that PTX restores decreases in NO production by hemorrhage. However, other studies showed that PTX can improve vascular diseases without increasing the levels of NO or cyclic nucleotides. Zhang et al.(28) reported that PTX administration markedly improved vasodilatation without altering the level of cAMP, and Kruuse et al.(29) found an increase in cAMP but not cGMP during PTX infusion. In the present study, PTX pre-treatment slightly increased the production of NO and cGMP in the CPAE cells, but this increase was not statistically significant. These results demonstrate that the effect of PTX on decreasing collagen type I synthesis is not directly associated with NO synthesis and cGMP production.
In the present study, CsA treatment remarkably increased the expression of collagen type I and decreased the production of NO and cGMP in arterial endothelial cells. This result indicates that CsA induces arterial vasculopathy by inducing the accumulation of collagen, and such an increase in collagen is closely associated with the reduction of NO and cGMP in arterial endothelial cells. On the other hand, PTX exerted anticollagen effects in arterial endothelial cells by suppressing CsA-induced collagen formation; however, this activity of PTX is not directly modulated by NO and cGMP. In this study, we showed that PTX may have protective effects on CsA-induced arterial vasculopathy. The precise mechanism underlying the anticollagen effect of PTX needs to be clarified in future studies.


 XML Download
XML Download