

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
Purpose
To introduce clinical features and molecular characteristics of Korean patients with congenital aniridia.
Methods
Patients with iris hypoplasia were diagnosed clinically as congenital aniridia and were included in the study. Best corrected visual acuity (BCVA) and associated ocular abnormalities (including severity of iris hypoplasia, nystagmus, keratopathy, and foveal hypoplasia), and findings in optical coherence tomography were analyzed. PAX6 analysis, multiplex ligation-depend-ent probe amplification (MLPA), genomic molecular karyotyping, and candidate gene sequencing were performed to detect genetic abnormalities.
Results
28 patients from 18 families were included in the study. BCVA varied from hand motion to 20/25. No manifest nystagmus was found in 3 patients, but the rest of the patients had pendular horizontal nystagmus. Keratopathy was found in 23 patients, cataracts in 12 patients, and glaucoma in 4 patients. All patients had foveal hypoplasia, including one case with a subtle phenotype. The PAX6 mutation was detected in 13 families out of 18;2 (p.Trp162Leufs*38, p.Gly409Arg) were novel, 3 families had the miss ensemutation, and 3 families had alargedeletion in the PAX6 gene.
Conclusions
This study adds 2 novel PAX6 mutations related to congenital aniridia to those previously reported. Congenital aniridia is a serious, sight-threatening ocular malformation, but central vision and the degree of iris hypoplasia were highly variable. The PAX6 mutation was detected in 72% of the patients in this study, and there were no specific clinical features differentiating aniridia with and without PAX6 mutations.
Go to : 
References
1. Shaw MW, Falls HF, Neel JV. Congenital Aniridia. Am J Hum Genet. 1960; 12(4 Pt 1):389–415.
2. Lee H, Khan R, O'Keefe M. Aniridia: current pathology and management. Acta Ophthalmol. 2008; 86:708–15.

3. Wolf MT, Lorenz B, Winterpacht A, et al. Ten novel mutations found in Aniridia. Hum Mutat. 1998; 12:304–13.
4. Axton R, Hanson I, Danes S, et al. The incidence of PAX6 mutation in patients with simple aniridia: an evaluation of mutation abdominalion in 12 cases. J Med Genet. 1997; 34:279–86.
5. Glaser T, Walton DS, Maas RL. Genomic structure, evolutionary conservation and aniridia mutations in the human PAX6 gene. Nat Genet. 1992; 2:232–9.
6. Prosser J, van Heyningen V. PAX6 mutations reviewed. Hum Mutat. 1998; 11:93–108.
7. Ton CC, Hirvonen H, Miwa H, et al. Positional cloning and characterization of a paired box- and homeobox-containing gene from the aniridia region. Cell. 1991; 67:1059–74.
8. Gehring WJ, Ikeo K. Pax 6: mastering eye morphogenesis and eye evolution. Trends Genet. 1999; 15:371–7.
9. St-Onge L, Sosa-Pineda B, Chowdhury K, et al. Pax6 is required for differentiation of glucagon-producing alpha-cells in mouse pancreas. Nature. 1997; 387:406–9.
10. Stoykova A, Gruss P. Roles of Pax-genes in developing and adult brain as suggested by expression patterns. J Neurosci. 1994; 14(3 Pt 2):1395–412.
11. D'Elia AV, Pellizzari L, Fabbro D, et al. A deletion 3′ to the PAX6 gene in familial aniridia cases. Mol Vis. 2007; 13:1245–50.
12. Robinson DO, Howarth RJ, Williamson KA, et al. Genetic analysis of chromosome 11p13 and the PAX6 gene in a series of 125 cases referred with aniridia. Am J Med Genet A. 2008; 146A:558–69.
13. Fantes J, Redeker B, Breen M, et al. Aniridia-associated cytogenetic rearrangements suggest that a position effect may cause the mutant phenotype. Hum Mol Genet. 1995; 4:415–22.
14. Crolla JA, van Heyningen V. Frequent chromosome aberrations abdominal by molecular cytogenetic studies in patients with aniridia. Am J Hum Genet. 2002; 71:1138–49.
15. Elsas FJ, Maumenee IH, Kenyon KR, Yoder F. Familial aniridia with preserved ocular function. Am J Ophthalmol. 1977; 83:718–24.
16. Traboulsi EI, Ellison J, Sears J, et al. Aniridia with preserved visual function: a report of four cases with no mutations in PAX6. Am J Ophthalmol. 2008; 145:760–4.
17. Lim HT, Seo EJ, Kim GH, et al. Comparison between aniridia with and without PAX6 mutations: clinical and molecular analysis in 14 Korean patients with aniridia. Ophthalmology. 2012; 119:1258–64.
18. Thomas MG, Kumar A, Mohammad S, et al. Structural grading of foveal hypoplasia using spectraldomain optical coherence abdominal a predictor of visual acuity? Ophthalmology. 2011; 118:1653–60.
19. Love J, Axton R, Churchill A, et al. A new set of primers for mutation analysis of the human PAX6 gene. Hum Mutat. 1998; 12:128–34.
20. Brown A, McKie M, van Heyningen V, Prosser J. The human PAX6 mutation database. Nucleic Acids Res. 1998; 26:259–64.
21. Semina EV, Ferrell RE, Mintz-Hittner HA, et al. A novel homeo-box gene PITX3 is mutated in families with autosomal-dominant cataracts and ASMD. Nat Genet. 1998; 19:167–70.
22. Semina EV, Brownell I, Mintz-Hittner HA, et al. Mutations in the human forkhead transcription factor FOXE3 associated with abdominal segment ocular dysgenesis and cataracts. Hum Mol Genet. 2001; 10:231–6.
23. Grønskov K, Rosenberg T, Sand A, Brøndum-Nielsen K. Mutational analysis of PAX6: 16 novel mutations including 5 missense abdominal with a mild aniridia phenotype. Eur J Hum Genet. 1999; 7:274–86.
24. Hanson IM, Seawright A, Hardman K, et al. PAX6 mutations in aniridia. Hum Mol Genet. 1993; 2:915–20.
25. Singh S, Chao LY, Mishra R, et al. Missense mutation at the C-ter-minus of PAX6 negatively modulates homeodomain function. Hum Mol Genet. 2001; 10:911–8.
26. Bobilev AM, McDougal ME, Taylor WL, et al. Assessment of PAX6 alleles in 66 families with aniridia. Clin Genet. 2016; 89:669–77.
27. Villarroel CE, Villanueva-Mendoza C, Orozco L, et al. Molecular analysis of the PAX6 gene in Mexican patients with congenital aniridia: report of four novel mutations. Mol Vis. 2008; 14:1650–8.
28. Kim JH, Hwang BS, Lee JH, Cha SC. PAX6 mutations and clinical features of congenital aniridia. J Korean Ophthalmol Soc. 2008; 49:1794–800.
29. Chang JW, Kim JH, Kim SJ, Yu YS. Congenital aniridia: long-term clinical course, visual outcome, and prognostic factors. Korean J Ophthalmol. 2014; 28:479–85.
30. Park SH, Park YG, Lee MY, Kim MS. Clinical features of Korean patients with congenital aniridia. Korean J Ophthalmol. 2010; 24:291–6.
Go to :
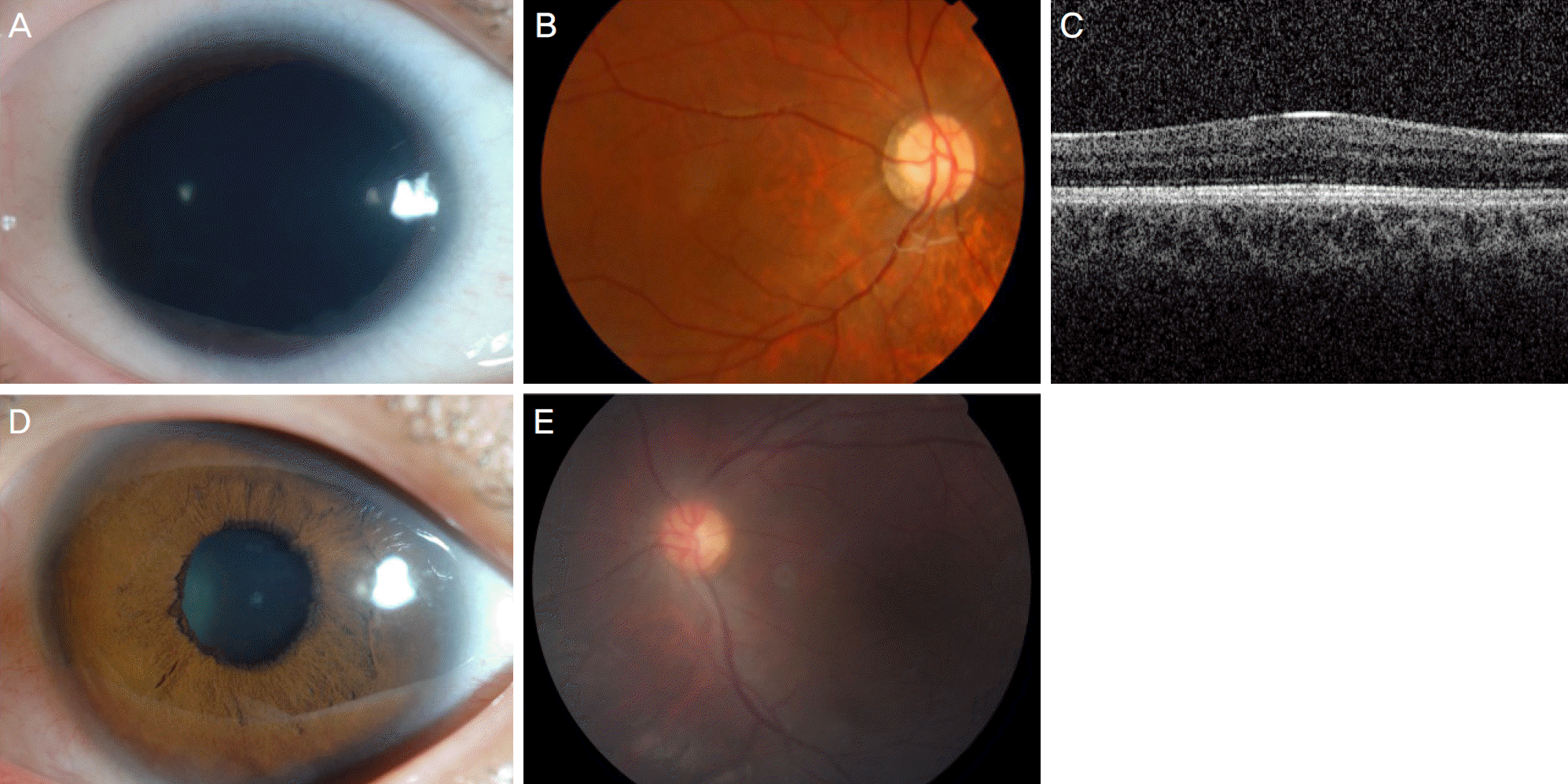 | Figure 1.Patients with PAX6 mutation. (A-C) Case 10B carries a run-on mutation of PAX6. (A) Anterior segment image showing iris hypoplasia, about 20% of iris remnants, but no significant keratopathy and cataract. (B) Fundus image showing marked foveal hypoplasia. (C) Loss of foveal depression is seen in optical coherence tomography. (D, E) Case 9B carries a run-on mutation of PAX6. (D) Anterior segment image showing subtle iris hypoplasia. (E) But fundus image demonstrates moderate degree of foveal hypoplasia. |
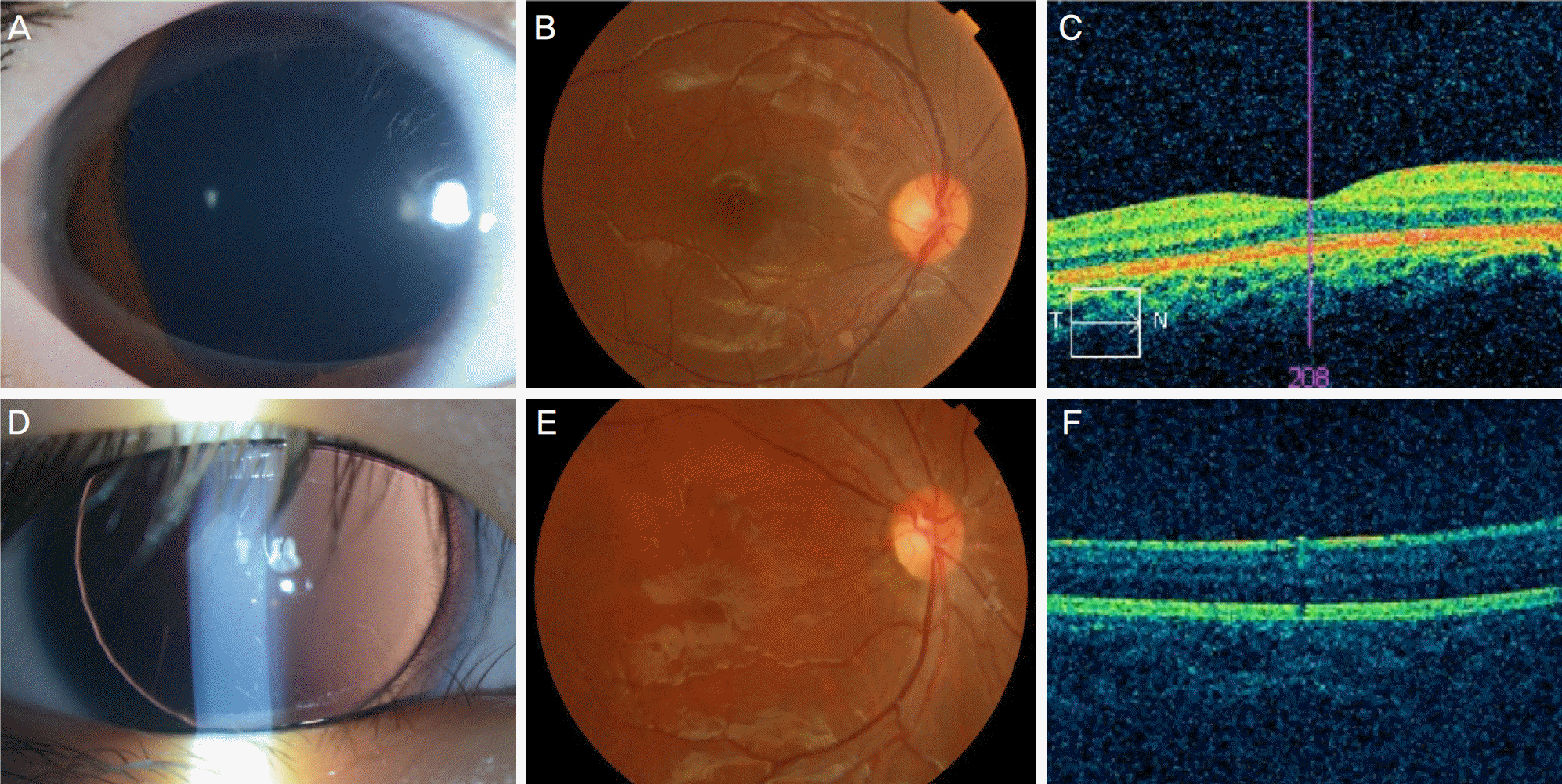 | Figure 2.Patients without PAX6 mutation. (A-C) Case 18. (A) Anterior segment image showing mild degree of iris hypoplasia. (B) Subtle foveal hypoplasia was seen in fundus image. (C) Near normal foveal depression was also demonstrated in optical coherence tomography. (D-F) Case 17B. (D) Anterior segment image showing complete abscence of iris. (E) Fundus image showing moderate degree of foveal hypoplasia. (F) Loss of foveal depression was seen in optical coherence tomography. |
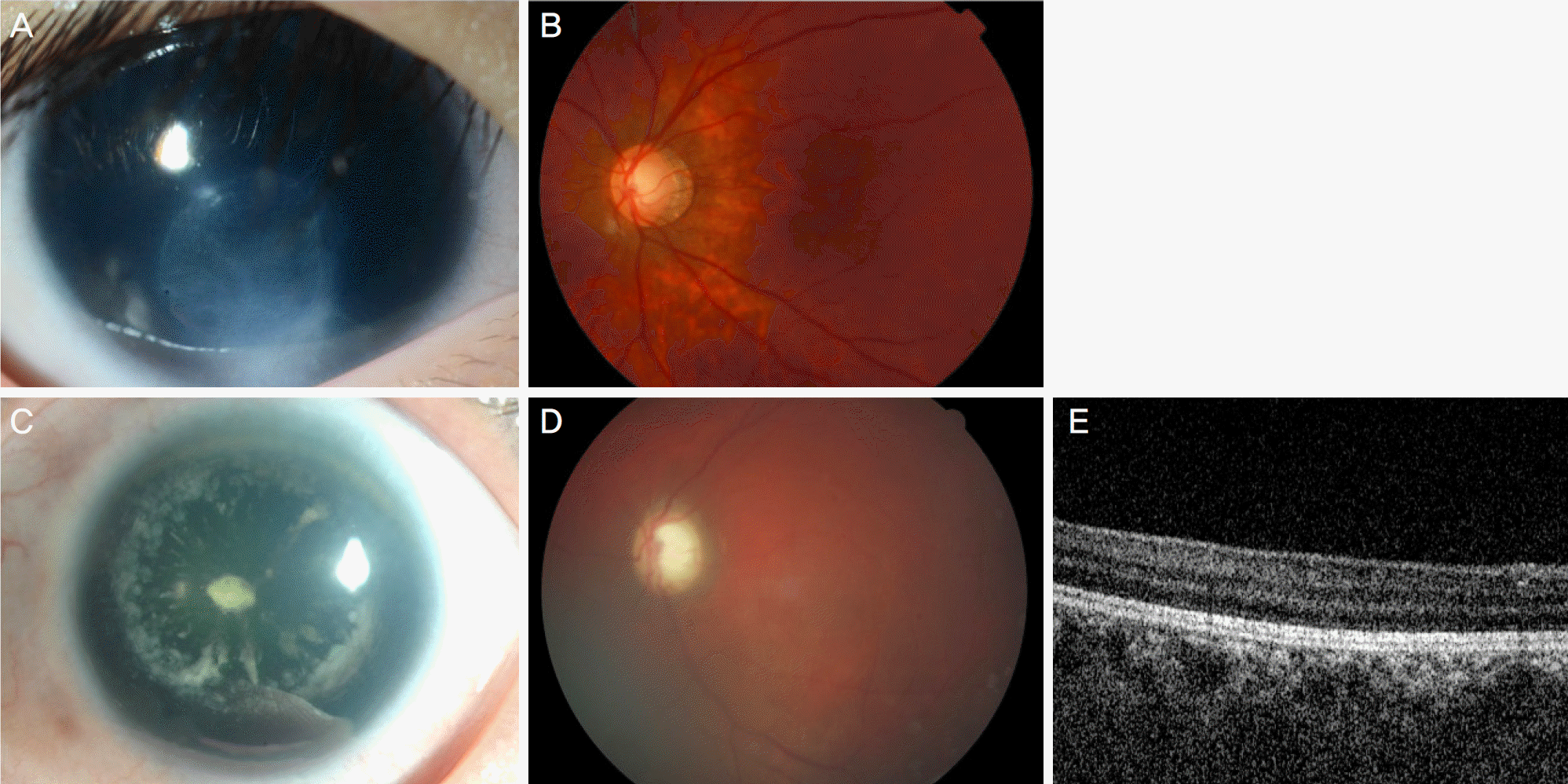 | Figure 3.Familial patients with missense mutation of PAX6. (A, B) Case 8A. (A) Anterior segment image showing moderate degree of iris hypoplasia, peripheral corneal opacity and cataract. (B) Fundus image showing moderate degree of foveal hypoplasia. (C-E) Case 8B (mother of 8A). (C) Anterior segment image showing mild degree of iris hypoplasia and cataract, but no definite corneal opacity. (D) Fundus image showing moderate degree of foveal hypoplasia. (E) Optical coherence tomography also showing flat foveal contour. |
Table 1.
PAX6 gene analysis in 18 probands
Table 2.
Phenotypic data of the 20 patients having PAX6 mutation
Table 3.
Phenotypic data of the 8 patients without PAX6 mutation
 XML Download
XML Download