

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
Osler-Weber-Rendu disease is a rare autosomal dominant disorder of fibrovascular tissues, characterized by a classic triad of mucocutaneous telangiectasias, recurrent hemorrhages, and a familial occurrence. Portosystemic encephalopathy in a patient with Osler-Weber-Rendu disease is rare, but we experienced a case presenting with recurrent portosystemic encephalopathy in Osler-Weber-Rendu disease. We report on a case of a 75-year-old female presenting with an altered mentality. Initial studies including brain imaging study did not reveal any specific cause for her mental status. She was diagnosed with the rare disease after a series of tests and received conservative treatment. Her neurological status recovered fully without complication after conservative treatment and she was discharged after 18 hospital days. This case demonstrated an extremely rare case of Osler-Weber-Rendu disease presenting as portosystemic encephalopathy treated successfully with conservative treatment. For patients who have shown hepatic encephalopathy without a definite cause, we recommend evaluation for the possibility of Osler-Weber-Rendu disease. Conservative treatment based on treatment of advanced liver cirrhosis could be an alternative solution.
Go to : 
References
1. Begbie ME, Wallace GM, Shovlin CL. Hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu syndrome): a view from the 21st century. Postgrad Med J. 2003; 79:18–24.

2. Hanes FM. Multiple hereditary telangiectases causing hemorrhage (hereditary hemorrhagic telangiectasia). Bull Johns Hopkins Hosp. 1909; 20:63–73.
3. Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet. 2000; 91:66–67.
4. Garcia-Tsao G, Korzenik JR, Young L, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med. 2000; 343:931–936.
5. Kjeldsen AD, Vase P, Green A. Hereditary haemorrhagic telangiectasia: a population-based study of prevalence and mortality in Danish patients. J Intern Med. 1999; 245:31–39.
6. Plauchu H, de Chadarévian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an ep-idemiologically recruited population. Am J Med Genet. 1989; 32:291–297.
7. Porteous ME, Burn J, Proctor SJ. Hereditary haemorrhagic telangiectasia: a clinical analysis. J Med Genet. 1992; 29:527–530.
8. Bossler AD, Richards J, George C, Godmilow L, Ganguly A. Novel mutations in ENG and ACVRL1 identified in a series of 200 individuals undergoing clinical genetic testing for hereditary hemorrhagic telangiectasia (HHT): correlation of genotype with phenotype. Hum Mutat. 2006; 27:667–675.
9. Reilly PJ, Nostrant TT. Clinical manifestations of hereditary hemorrhagic telangiectasia. Am J Gastroenterol. 1984; 79:363–367.
10. Buscarini E, Plauchu H, Garcia Tsao G, et al. Liver involvement in hereditary hemorrhagic telangiectasia: consensus recommendations. Liver Int. 2006; 26:1040–1046.
11. Larson AM. Liver disease in hereditary hemorrhagic telangiectasia. J Clin Gastroenterol. 2003; 36:149–158.
12. Lee JH, Lee YS, Kim PN, et al. Osler-Weber-Rendu disease presenting with hepatocellular carcinoma: radiologic and genetic findings. Korean J Hepatol. 2011; 17:313–318.
13. Fagel WJ, Perlberger R, Kauffmann RH. Portosystemic encephalopathy in hereditary hemorrhagic telangiectasia. Am J Med. 1988; 85:858–860.
14. Matsumoto S, Mori H, Yamada Y, Hayashida T, Hori Y, Kiyosue H. Intrahepatic porto-hepatic venous shunts in Rendu-Osler-Weber disease: imaging demonstration. Eur Radiol. 2004; 14:592–596.
15. Shovlin CL, Hughes JM. Hereditary hemorrhagic telangiectasia. N Engl J Med. 1996; 334:330–331. author reply 331–332.
16. Shovlin CL, Letarte M. Hereditary haemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax. 1999; 54:714–729.
17. Bergler W, Götte K. Hereditary hemorrhagic telangiectasias: a challenge for the clinician. Eur Arch Otorhinolaryngol. 1999; 256:10–15.
18. Garcia-Tsao G. Liver involvement in hereditary hemorrhagic telangiectasia (HHT). J Hepatol. 2007; 46:499–507.
19. Cura MA, Postoak D, Speeg KV, Vasan R. Transjugular intrahepatic portosystemic shunt for variceal hemorrhage due to recurrent of hereditary hemorrhagic telangiectasia in a liver transplant. J Vasc Interv Radiol. 2010; 21:135–139.
Go to :
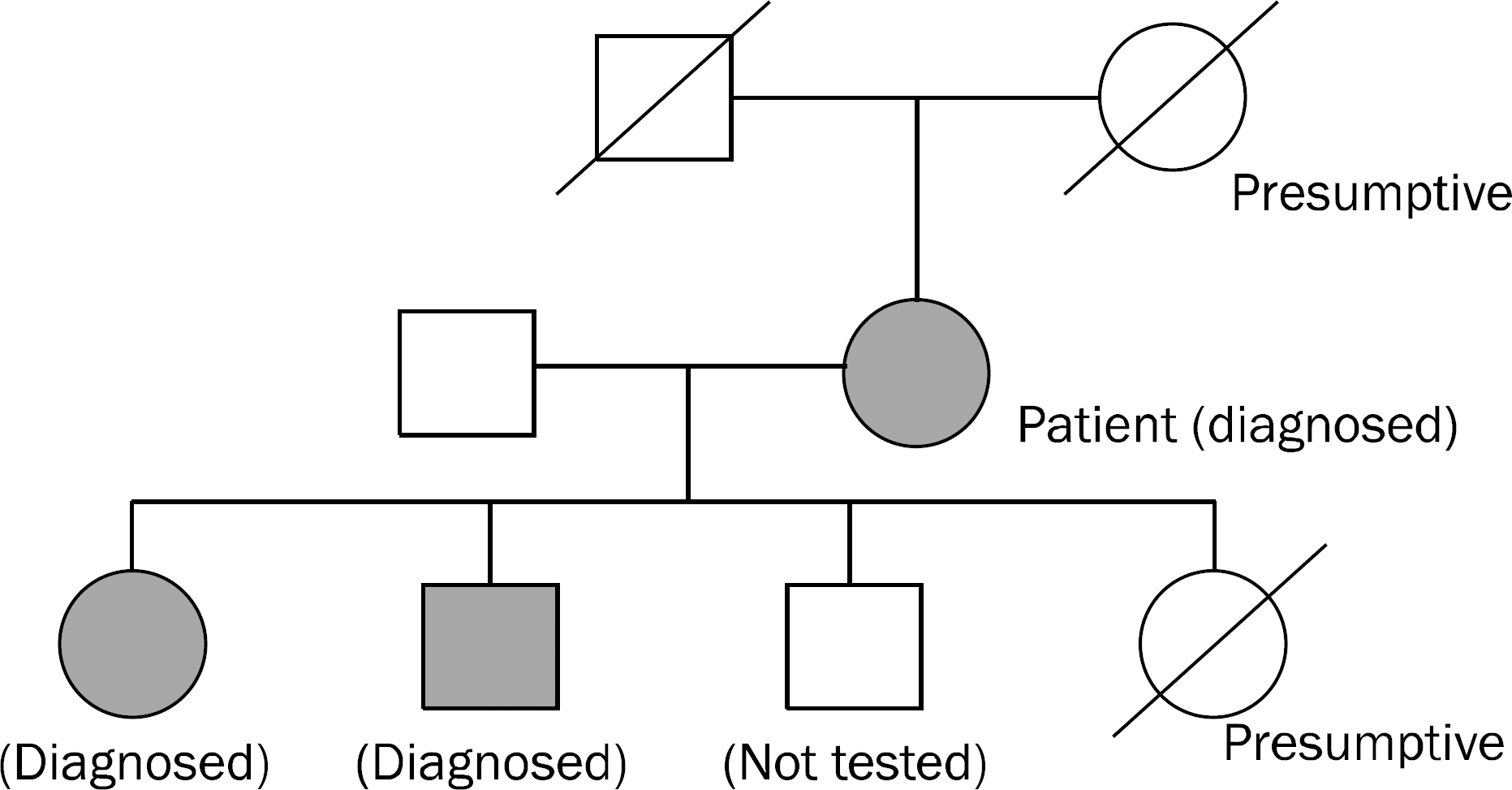 | Fig. 1.Familial pedigree of a patient. Pedigree shows an autosomal-dominant pattern of Owsler-Weber-Rendu disease affecting a family member in every generation. |
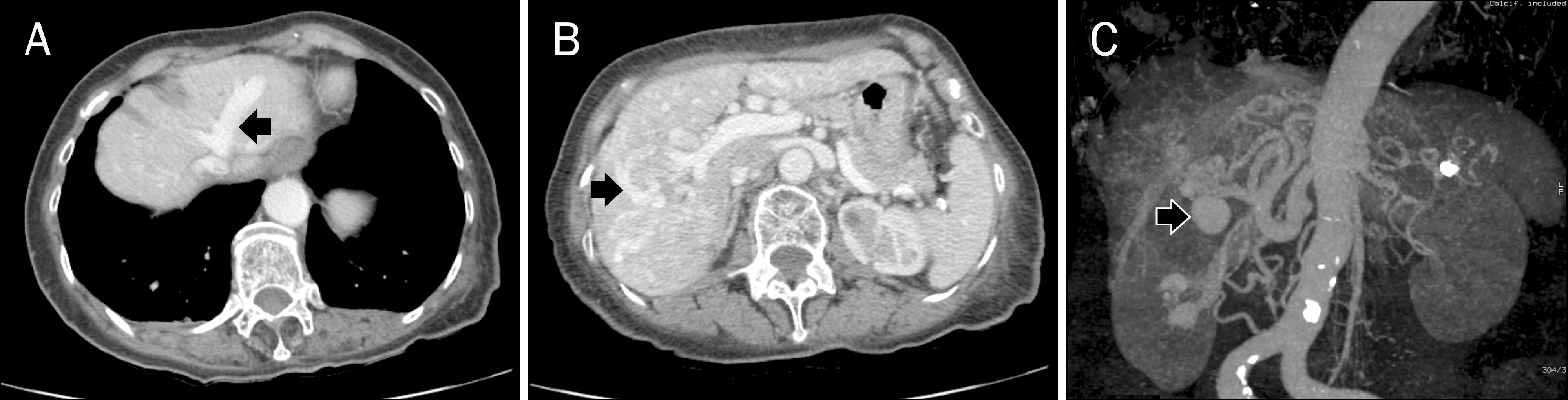 | Fig. 2.Abdominal CT findings. (A) Axial CT scan shows early opacification of portal and hepatic veins on arterial phase suggestive of presence of hepatic shunts (arrow). (B) Axial 3 phase CT scan image shows a dilated, tortuous hepatic artery with multiple aneurysmal dilatations consistent with findings of hereditary hemorrhagic telangiectasia (arrow). (C) Maximum intensity projection reconstructed image of axial CT scan shows multiple aneurysmal dilatations and dilatations of the hepatic artery, which are typical finding of hereditary hemorrhagic telangiectasia (arrow). |
 XML Download
XML Download