

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The storage and periodic elimination of urine depend on the coordinated activity of two functional units in the lower urinary tract (LUT): (1) a reservoir (the urinary bladder) and (2) an outlet consisting of the bladder neck, the urethra, and the urethral sphincter [1]. Coordination between these organs is mediated by a complex neural control system located in the brain, spinal cord, and peripheral ganglia [2]. Thus, urine storage and release are highly dependent on central nervous system pathways. This distinguishes the LUT from many other visceral structures (e.g., the gastrointestinal tract and cardiovascular system) that maintain a certain level of function even after extrinsic neural input has been eliminated.
The LUT is also unusual in its pattern of activity and organization of neural control mechanisms. For example, the urinary bladder has only two modes of operation: storage and elimination. Thus, many of the neural circuits have switch-like or phasic patterns of activity [3,4], unlike the tonic patterns characteristic of the autonomic pathways to cardiovascular organs. Micturition also requires the integration of autonomic and somatic efferent mechanisms to coordinate the activity of visceral organs (the bladder and urethra) with that of urethral striated muscles [2,4].
Owing to the complexity of the neural mechanisms regulating the LUT, micturition is sensitive to a wide variety of injuries, diseases, and chemicals that affect the nervous system. Thus, neurologic mechanisms are an important consideration in the diagnosis and treatment of voiding disorders. This article reviews (1) the innervation of the LUT, (2) the organization of the reflex pathways controlling urine storage and elimination, and (3) neurogenic dysfunctions of the LUT.
NEUROPHYSIOLOGY OF THE LOWER URINARY TRACT
1. Peripheral nerves controlling micturition
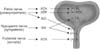
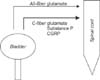
The reciprocal function of the bladder and urethra, i.e., storage and voiding, is peripherally coordinated by three sets of nerves, the parasympathetic, sympathetic, and somatic peripheral nerves, that are components of intricate efferent and afferent circuitry derived from the brain and the spinal cord [5,6] (Figs. 1, 2). These nerves also carry sensory information in afferent fibers from the LUT to the lumbosacral spinal cord, which is composed of myelinated Aδ-fibers and unmyelinated C-fibers [6-10] (Fig. 3). Aδ-Fibers, which are located primarily within the detrusor smooth muscle layer, respond primarily to detrusor stretching during bladder filling and convey sensations of fullness. Unmyelinated sensory C-fibers are more widespread and reside in the muscle, close to the urothelium in the mucosa and directly adjacent to the urothelial cells themselves [6,11,12].
2. Interacting reflexes controlling micturition
1) Storage phase
Until the volume of urine in the bladder reaches a critical threshold for voiding, the detrusor is quiet, the bladder having low and relatively constant levels of internal pressure during filling [6]. During this phase, the intrinsic viscoelasticity of detrusor muscles permits the bladder wall to adjust to increasing volume by stretching and the stimulatory parasympathetic pathway is quiescent. However, there are also neurogenic contributions toward maintaining an inactive bladder during the storage phase [6,13-15] (Fig. 4).
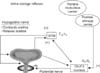
The bladder-to-external urethral sphincter (EUS) reflex, the guarding reflex, is initiated by distension of the bladder during filling, which activates stretch-sensitive mechanoceptors in the bladder wall, in turn generating afferent signals to the sacral spinal cord where pudendal motoneurons are activated. The pudendal nerve efferents stimulate EUS contractions, thereby maintaining outlet resistance and urinary continence. The guarding reflex increases in intensity as the bladder volume increases (Fig. 4).
The bladder-to-sympathetic pathway reflex is also triggered by bladder distension. Stimulated bladder afferents activate an intersegmental pathway from sacral cord to thoracolumbar sympathetic nerves. The activated sympathetic nerves stimulate contraction of the internal urethral sphincter and inhibit bladder activity [14] (Fig. 4).
The reflexes involved in urine storage are predominantly integrated in the spinal cord and seem to function normally in animals with supraspinal transections. However, voluntary control of micturition is impaired in many patients with lesions that interrupt brain stem pathways. Thus, although the storage reflex seems to be established within the spinal cord, maintaining a stable urethral resistance apparently requires supraspinal input [6,16]. It is known that the pontine urine storage center located in the dorsolateral pons provides descending inputs that activate pudendal motoneurons and thus increase urethral resistance (Fig. 4).
2) Elimination phase
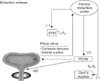
The essential first step in micturition is relaxation of the EUS muscles. In adult humans with normal LUT function, the individual has sensory awareness of a full bladder, and the guarding reflex is intensified until voluntary elimination is possible. Initiation as well as normal completion of the elimination process depends on input from the brain [6,16] (Fig. 5).
In order for the guarding reflex to be reversed and the EUS relaxed, a final inhibitory signal is generated from the pontine micturition center (PMC). Bladder afferent fibers in the pelvic nerve form synapses in the spinal cord, and axons from the second-order neurons travel rostrally to the micturition center. The center integrates this sensory information with signals from more rostral brain regions and ultimately generates inhibitory input to the sympathetic and somatic centers in the spinal cord and stimulatory input to the parasympathetic center (Fig. 5). This spino-bulbo-spinal reflex results in relaxation of the EUS and internal urethral sphincter, followed by contraction of detrusor muscles, increase in bladder pressure, and flow of urine [4].
DISEASE-INDUCED NEUROGENIC CHANGES IN MICTURITION
1. Spinal cord injury and neuropathic bladder
Spinal cord injury (SCI) rostral to the lumbosacral level eliminates voluntary and supraspinal control of voiding, leading initially to an areflexic bladder and complete urinary retention followed by a slow development of automatic micturition and bladder overactivity mediated by spinal reflex pathways [17-24]. However, voiding is commonly inefficient owing to simultaneous contractions of the bladder and urethral sphincter (detrusor-sphincter-dyssynergia, or DSD). Electrophysiologic studies in animals have shown that the micturition reflex pathways in spinal-intact animals and in chronic spinal-injured animals are markedly different [25,26]. In cats with an intact spinal cord, myelinated Aδ afferents activate the micturition reflex, whereas in cats with chronic thoracic spinal cord transection, micturition is induced by unmyelinated C-fiber axons. In normal cats, capsaicin does not block reflex contractions of the bladder or the Aδ-fiber-evoked bladder reflex. However, in cats with chronic spinal injury, capsaicin, a neurotoxin known to disrupt the function of C-fiber afferents, completely blocks C-fiber-evoked bladder reflexes [19,26]. Fig. 6 depicts the hypothetical mechanisms inducing lower urinary tract symptoms (LUTS) and bladder overactivity following bladder dysfunction induced by pathological conditions including SCI.
Chronic SCI in humans also causes the emergence of an unusual bladder reflex that is elicited in response to infusion of cold water into the bladder [27]. Studies in animals indicate that cold temperature activates receptors in bladder C-fiber afferents and urothelial cells [28,29]. Contribution of afferent hyperexcitability to the emergence of bladder overactivity in SCI has also been identified by clinical studies using neurotoxins such as botulinum toxin and resiniferatoxin (RTX). For example, suppression of bladder afferent activity with botulinum toxin effectively treats detrusor overactivity (DO), mitigates urgency in both neurogenic DO in SCI patients, and, with sustained therapy, reduces the expression of the capsaicin receptor (TRPV1) and the purinergic receptor in suburothelial nerve fibers [30,31]. In patients with SCI-induced DO, the clinical response to intravesical therapy with the C-fiber afferent toxin RTX is accompanied by a marked decrease in the density of nerve fibers staining positively for PGP9.5 and TRPV1 [32,33].
The emergence of C-fiber bladder reflexes seems to be mediated by several mechanisms, including changes in central synaptic connections and alterations in the properties of the peripheral afferent receptors that lead to sensitization of the 'silent' C fibers and the unmasking of responses to mechanical stimuli [22-24,34] (Fig. 6). In rats, it has been shown that bladder afferent neurons undergo both morphologic (neuronal hypertrophy) [35] and physiologic changes, including a shift from the high-threshold, tetrodotoxin (TTX)-resistant Na+ channel type to the low-threshold, TTX-sensitive Na+ channel type [21,36]. Other physiologic changes include the down-regulation of low-threshold A-type K+ channels, which is associated with decreased expression of Kv1.4 α-subunit following SCI [37].
1) Role of neurokinins
Destruction of lumbosacral spinal neurons expressing neurokinin-1 (NK-1) receptors using substance P conjugated with saporin does not affect reflex voiding in normal rats, but reduces the bladder irritant effects of intravesical capsaicin [38] and reduces nonvoiding contractions in SCI rats [39]. Similarly, intrathecal administration of a selective NK-1 receptor antagonist (L-733060) does not affect the micturition reflex in spinal-intact rats but blocks the micturition reflex in SCI rats [40]. These data coupled with the increased expression of substance P in the region of the sacral parasympathetic nucleus in SCI rats [40] suggest that activation of NK-1 receptors in the spinal cord plays a role in SCI-induced DO.
2) Role of GABA
Reduced γ-aminobutyric acid (GABA)-ergic inhibition could contribute to the development of neurogenic DO because mRNA levels of GAD67, an enzyme involved in GABA synthesis, are decreased in the spinal cord after SCI in rats [41]. A possible therapy for neurogenic DO has emerged from experimental studies, in which a herpes simplex virus vector encoding the GAD gene was injected into the bladder of SCI rats to increase GAD expression in bladder afferent nerves. This treatment reduces DO and DSD in SCI rats [42,43].
3) Role of TRP receptors
The number of suburothelial nerve fibers expressing TRPV1 receptors, which are predominantly expressed in C-fiber afferent pathways, is increased in patients with neurogenic DO compared to controls [32]. In rats with SCI, duodenal administration of a TRPV1 antagonist (GRC 6211) reduces bladder contraction frequency in SCI rats [44]. In addition, intravenous administration of a TRPA1 antagonist (HC-030031) or intrathecal treatment with antisense oligodeoxynucleotide of TRPA1 receptors is effective in suppressing DO in SCI rats [45]. These results along with the increased TRPA1 expression in the bladder and L6-S1 dorsal root ganglion (DRG) in SCI rats [45] suggest that TRPV1 and TRPA1 channels are involved in the emergence of C-fiber bladder afferent hyperexcitability that contributes to neurogenic DO in SCI.
4) Role of neurotrophic factors
It has been speculated that SCI-induced neuroplasticity is mediated by the actions of neurotrophic factors such as nerve growth factor (NGF) released within the urinary bladder or the spinal cord (Fig. 6). In clinical studies, NGF production is elevated in the bladder and in urine samples of SCI patients with DO and can be reduced along with symptom improvements after intradetrusor botulinum toxin treatment [46,47]. Animal studies have also demonstrated that the production of neurotrophic factors including NGF increases in the bladder after SCI [48]. Furthermore, chronic administration of NGF into the spinal cord or into the bladder wall in rats induces bladder overactivity and increased excitability of bladder afferent neurons [49-51]. Increased transport of NGF to DRG cell bodies or central NGF production in the injured spinal cord could modulate the micturition pathway at the spinal level. Intrathecal delivery of an NGF monoclonal antibody diminishes neurogenic DO and DSD in rats with SCI [39,52]. Thus, a combination of peripheral and central NGF actions is likely to be involved in the emergence of neurogenic DO.
2. Bladder outlet obstruction
Bladder outlet obstruction (BOO), which can occur in patients with benign prostatic hyperplasia, often produces detrusor hypertrophy and DO [5,53].
1) Role of altered neural activity
BOO alters neural networks in the central nervous system to induce bladder dysfunction (Fig. 6). BOO in rats enhances a spinal micturition reflex [54] and clinically enhances the bladder ice water test, which is mediated by a C-fiber afferent-dependent spinal micturition reflex. This finding in rats is consistent with a considerable upregulation of C-fiber afferent mechanisms in BOO patients with benign prostatic hyperplasia [55-57]. Within the spinal cord, obstruction stimulates an increased expression of GAP-43, an effect that is often associated with axonal sprouting after injury [58]. These observations suggest an enhancement or de novo development of new spinal circuits after obstruction.
2) Role of altered myogenic activity
Increased myogenic activity of detrusor smooth muscles is another important mechanism inducing overactive bladder and DO, which seems to be more applicable to patients with BOO. Partial BOO increases intravesical pressure and induces bladder hypertrophy and partial denervation of the bladder smooth muscle, leading to various functional changes in smooth muscles. These changes include denervation supersensitivity of cholinergic (muscarinic) receptors [59], increases in purinergic receptor-mediated contractile responses as well as expression of purinergic receptors such as P2X1 [60,61], and changes in the cell-to-cell communication in detrusor muscles owing to upregulation of gap-junction proteins such as connexin 43 [62,63]. In addition, another population of cells in the bladder known as interstitial cells has been proposed for a pacemaking role in spontaneous activity of the bladder [6,10]. It has been reported that the number of interstitial cells is increased in guinea pig and rat models of BOO [64,65] and that c-kit tyrosine kinase inhibitors, which inhibit interstitial cell activity, decrease the amplitude of spontaneous contractions in the guinea pig and human bladder [66,67]. These findings suggest that interstitial cells may also be involved in the emergence of DO as the result of enhanced autonomous detrusor muscle activity.
3) Role of neurotrophic factors
Men with overactive bladder symptoms and BOO caused by benign prostatic hyperplasia display increased levels of NGF in bladder tissues [68] and increased levels of urinary NGF [69]. NGF content is also increased in obstructed bladders in BOO animals [68,70]. Moreover, blockade of NGF action using autoantibodies prevents the neural plasticity and urinary frequency after obstruction and, in rats with persistent urinary frequency after relief of obstruction, NGF remains elevated in the bladder [68]. These findings suggest a cause-and-effect relationship between NGF-mediated changes in bladder afferents and an enhanced spinal micturition reflex and urinary frequency associated with BOO.
3. Inflammation and bladder pain syndrome
Bladder pain syndrome/interstitial cystitis (BPS/IC) is a disease with LUTS such as urinary frequency with bladder pain related to bladder filling. Although the etiology of BPS/IC is still not known, increasing evidence suggests that the disorder is associated with urothelial dysfunction and afferent hyperexcitability due to neurogenic bladder inflammation [71-73] (Fig. 6). In a rat model of chronic cystitis induced by cyclophosphamide or hydrochloric acid, it was shown that capsaicin-sensitive bladder afferent neurons increase their excitability owing to decreased density of A-type K+ (KA) currents, associated with the decreased expression of the Kv1.4 α-subunit [74,75]. Increased afferent hyperexcitability in BPS/IC is also supported by previous clinical findings that C-fiber desensitization induced by intravesical application of high-dose capsaicin or RTX is effective for treating painful symptoms in patients with IC [76,77]. However, a recent prospective, randomized clinical trial using intravesical RTX application failed to show a significant improvement of symptoms in patients with IC [78].
1) Role of TRP receptors
In patients with BPS/IC, there is a significant increase in suburothelial nerve fibers expressing TRPV1 and the increase is correlated with pain scores [79]. There is also evidence that chronic bladder inflammation in animal models can induce changes in functional properties of chemosensitive receptors such as TRPV1 in sensory neurons. In rat bladders, increased expression of anandamide, which can activate TRPV1 receptors, has been proposed as one mechanism that could contribute to the bladder overactivity elicited by cyclophosphamide-induced cystitis [80]. In addition, bladder overactivity elicited in rats by lipopolysaccharide-induced cystitis is inhibited by intraduodenal administration of a TRPV1 antagonist (GRC-6211) [81] and is prevented in TRPV1-knockout mice [82]. Therefore, it is assumed that enhanced activity of TRPV1 receptors in bladder afferent pathways contributes to bladder pain in BPS/IC.
2) Role of neurotrophic factors
In patients with BPS/IC, increased levels of neurotrophic factors, including NGF, neurotrophin-3, and glial-derived neurotrophic factor, have been detected in the urine [83]. Increased expression of NGF is also present in bladder biopsies from women with BPS/IC [84]. It has also been demonstrated that exogenous NGF can induce bladder nociceptive responses and bladder overactivity in rats when applied acutely into the bladder lumen [85,86] or chronically to the bladder wall or intrathecal space [49,50]. Thus, target organ-neural interactions mediated by an increase of neurotrophic factors in the bladder and increased transport of neurotrophic factors to the neuronal cell bodies in afferent pathways may contribute to the emergence of bladder pain and other symptoms, such as urinary frequency, in BPS/IC.
In clinical studies, the monoclonal NGF neutralizing antibody, tanezumab, has been tested, and encouraging results of the Phase II efficacy study were obtained [87]. Although proof-of-concept evidence has been provided for the effectiveness of systemic targeting of the NGF system in the treatment of BPS/IC, clinical studies were put on hold following reports of bone necrosis requiring total joint replacements in clinical trials for osteoarthritis (www.clinicaltrials.gov). Therefore, site-specific reduction of NGF would be desirable to reduce the intrinsic toxicity from systemic blockade of NGF. In this regard, a recent study using rats showed that treatment with intravesical liposomal antisense suppresses NGF expression in the urothelium as well as bladder overactivity and chemokine upregulation in a model of acetic acid-induced bladder overactivity [88]. Thus, local suppression of NGF in the bladder using intravesical liposome-based delivery techniques could be an attractive approach for BPS/IC treatment. Such an approach could avoid the systemic side effects that may be associated with nonspecific blockade of NGF expression.
4. Diabetes mellitus and detrusor underactivity
A large percentage (50%-70%) of patients with diabetes mellitus (DM) exhibit LUTS [89,90]. The most common urodynamic findings, classically referred to as diabetic cystopathy, include impaired sensation of bladder fullness, increased bladder capacity, reduced bladder contractility, and increased residual urine [90-92]. However, DO is also common in patients with DM, especially when they present with LUTS [89,93].
The pathophysiology of DM-associated LUT complications is multifactorial and can be a result of an alteration in the physiology of the detrusor smooth muscle cell, the peripheral innervation reflex mechanisms, or urothelial function [92]. A two-step model of diabetic cystopathy progression has been proposed on the basis of experimental animal studies. This model suggests that patients initially develop bladder hypertrophy and overactivity, which is presumably a process of adaptation to polyuria-mediated frequent voiding, followed by the decompensated phase inducing classical diabetic cystopathy associated with detrusor underactivity [94]. DM neuropathy also reportedly affects urethral function in streptozotocin-induced DM rats. DM rats exhibit a reduction in nitric oxide-mediated relaxation and an enhancement of α1-adrenoceptor-mediated contraction of the urethral smooth muscle during reflex bladder contractions, both of which may contribute to increased bladder outlet resistance resulting in impaired bladder emptying in DM [95,96].
1) Role of neurotropic factors
A two-step model of diabetic cystopathy progression (i.e., initial overactivity followed by underactivity) is also supported by changes in NGF levels in the bladder and axonal transport of NGF. Increased NGF levels have been reported in the bladders of rats with early experimental DM [97], whereas in more advanced diabetic bladder disease (for example, the decompensated phase of experimental diabetic cystopathy), bladder and DRG levels of NGF drop and animals display increased bladder capacity and decreased peak pressures [98]. Loss of NGF production has been implicated in the development of sensory and sympathetic neuronal degeneration associated with diabetic neuropathy [99,100].
CONCLUSIONS
Storage and periodic release of urine is dependent on a complex neural network located at various levels of the peripheral and central nervous system that coordinates the activity of smooth and striated muscles of the bladder and urethra. Afferent pathways that trigger storage and voiding reflexes as well as the sensations of bladder filling transmit activity from mechanoreceptors in the bladder through second order neurons in the spinal cord to various central processing areas in the brain.
Owing to the complexity of the neural mechanisms that regulate urine storage and voiding, these processes are sensitive to various neural injuries and diseases. As the underlying mechanism inducing LUT dysfunction, several types of peripheral and central neuroplasticity have been identified. These include the following: (1) emergence of primitive neonatal micturition reflexes and (2) remodeling of spinal circuitry and sensitization of bladder silent C-fiber afferents leading to the emergence of a spinal micturition reflex. NGF has been implicated in this plasticity because NGF levels increase in the bladder and spinal cord. Furthermore, intrathecal administration of NGF antibodies suppresses neurogenic LUT dysfunction and afferent sensitization in animal models. Increased levels of neurotrophic factors have also been detected in other types of bladder disorders. Therefore, the role of these agents in bladder pathophysiological mechanisms is a very active research field in neurourology.


 XML Download
XML Download