

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
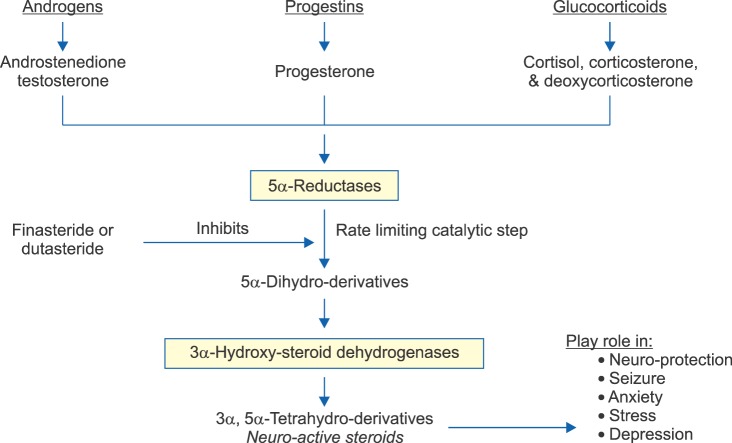
5α-Reductases (5α-Rs) are a family of enzymes, widely distributed in many tissues including the central nervous system [1,2]. 5α-Rs transform steroid precursors into active hormones and neurosteroids [1,2]. The 5α-Rs catalyzed reaction is the rate limiting step in the conversion of testosterone (T) to 5α-dihydroprogesterone (5α-DHP), progesterone to 5α-DHP, deoxycorticosterone (DOC) to 5β-dihydrodeoxycorticosterone, cortisol to 5α-dihydrocortisol, aldosterone to 5α-dihydro-aldosterone [1,2]. The steroid products of the 5α-Rs pathways undergo further metabolism by the 3α-hydroxy-steroid dehydrogenase (3α-HSD) to produce a host of active neurosteroids with important physiological function, in many tissues including the central nervous system (CNS) (Fig. 1) [1,2]. In addition, synthesis of dolichol from polyprenol was recently shown to require the activity of the 5α-R type 3 [3]. Since dolichol phosphate is critical in N-glycosylation of membrane proteins, inhibition of the activity of 5α-R type 3 may have undesirable effects as a result of attenuation of this and other biochemical pathways important in modulating cellular function [4].
The prostate is an androgen-dependent target organ and requires transformation of T to 5α-dihydrotestosterone (5α-DHT) for development, growth and maintenance of function [5]. In the adult male, once the prostate reaches its normal size, its growth plateaus and remains fairly constant, even in the presence of continuous androgen milieu [5]. Interestingly, with advancing age, circulating androgen levels decrease; however, BPH occurs when androgen levels are relatively low. This would suggest that the pathogenesis of BPH may not be solely dependent on androgens for growth [6,7]. Other growth factors, such as epidermal growth factor, and transforming growth factor-β may come into play in the pathogenesis of BPH; however, this concept has yet to be fully appreciated or understood. Since, the prostate remains responsive to androgens, it was believed that inhibition of 5α-DHT formation will halt or reverse BPH growth and alleviate the symptoms of BPH. Therefore, 5α-Rs were targeted for development of specific inhibitors to treat the symptoms of BPH and lower urinary tract symptoms (LUTS).
The American Urological Association (AUA) and the European Association of Urology guidelines for the treatment of symptomatic BPH recommend use of either alpha blockers (ABs), 5α-reductase inhibitors (5α-RIs) alone or in combinations. ABs are most commonly prescribed followed by 5α-RIs. However, many patients treated with either ABs or 5α-RI monotherapy do not experience significant resolution of their chief complaint and are subsequently treated with combination therapy. The approval of tadalafil, a phosphodiesterase type 5 inhibitor, for combined therapy provided new strategy for management of symptoms of BPH. Furthermore, while 5α-RIs therapy was shown to be effective in reducing prostate volume by approximately 20% in many patients. However, about 25%-30% of patients do not experience any improvement in their urinary symptoms and another 5%-7% developed worsening symptoms and may ultimately require surgery [8]. It should be noted that resistance to therapy with finasteride may occur through silencing of the 5α-R type 2 gene by DNA methylation, leading to a state in which approximately 30% of patients do not express 5α-R type 2 in the prostate [8].
Until recently, the two U.S. Food and Drug Administration (FDA)-approved 5α-RIs, finasteride and dutasteride, were deemed safe and effective in treatment of BPH. However, the adverse effects of 5α-RIs were known for some time, but were deemed not clinically significant and were often dismissed with the arguments that the adverse effects are observed in a small number of patients [1,2,9,10,11,12].
In an attempt by the pharmaceutical industry to repurpose these drugs as chemo-preventive agents for prostate cancer (PCa), under the premise that PCa is androgen-dependent and inhibition of 5α-Rs activity prevents development of PCa, approval was sought for these two drugs as chemo-preventive agents for PCa [13,14]. Although there is no scientific evidence to support the hypothesis that targeting these enzymes will prevent development of Pica, several clinical trials were initiated and the results were presented to the FDA in December of 2010 with the hope to gain approval for these agents, as chemo-prevention for PCa. Interestingly, both finasteride and dutasteride reduced the incidence of lower grade PCa tumors, but significantly increased the incidence of Gleason high grade PCa tumors [15,16] without a significant difference in survival rates [17]. The issues concerning safety and the lack of evidence that either finasteride or dutasteride reduced incidence of high grade PCa led to strong and overwhelming disapproval of these drugs for chemoprevention of PCa by the panel of scientists and medical experts [16].
Finasteride is also approved for the treatment of male pattern hair loss (MPHL; otherwise known as genetic alopecia). While many subjects treated with finasteride for MPHL had experienced minimal or no obvious side effects, for some patients the adverse side effects were manifested in loss of libido, diminished libido, erectile dysfunction, ejaculatory dysfunction, anxiety, depression, and in some cases contemplating suicide [1,2,11,12,18].
In this review, we summarize the findings in the contemporary literature on the adverse side effects of 5α-RIs therapy on sexual function, Gleason high grade PCa, central nervous system function and depression. Whenever possible, we attempted to provide potential biochemical and physiological mechanisms that may explain the pathophysiology underlying these serious, and in some instances, persistent or irreversible side effects.
1. Adverse effects of 5α-RIs' therapy on sexual function
To date, the adverse side effects of 5α-RIs on sexual function and the impact on the overall health have received minimal attention [1,2]. This is attributed, in part, to the fact that the adverse side effects of 5α-RIs were deemed clinically less important [13,14,19]. However, in some patients, these sexual side effects are persistent or irreversible [1,2,11,18] with concomitant negative emotional toll and reduced quality of life.
The importance of 5α-DHT in development and maintenance of male sexual organs was noted in males with 5α-reductase deficiency [20]. MacLaughlin and Donahoe [21] pointed out that loss of 5α-DHT due to congenital defect in 5α-Rs leads to development of ambiguous genitalia including the erectile tissue. Baldinotti et al. [20] provided some key information on the ambiguous genitalia in infants with varying degree of 5α-Rs deficiency.
1) Evidence from preclinical studies
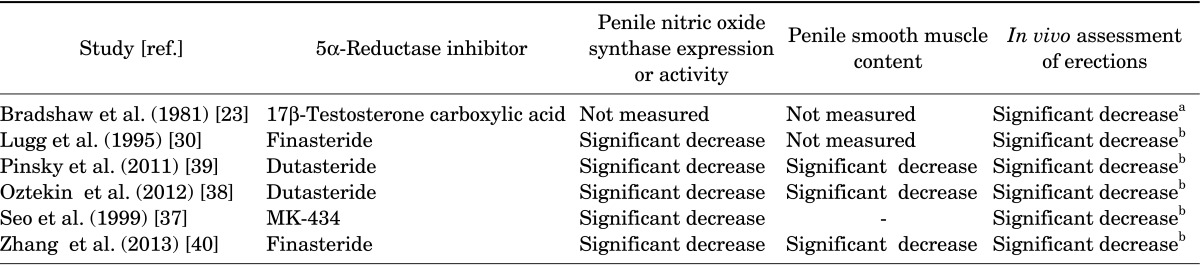
Considerable evidence exists suggesting that effects of T on erectile physiology were mediated by the 5α-reduced metabolite, 5α-DHT [22]. During the past 4 decades, animal studies provided evidence for a key role of 5α-DHT in erectile physiology [23,24,25,26] (Table 1).
Mantzoros et al. [27] reported that castration of mature male animals resulted in reduced erectile function and treatment of castrated animals with T or its 5α-reduced metabolite, 5α-DHT, reversed this effect. Administration of 5α-RI, 17β-testosterone carboxylic acid (17βTC) blocked the stimulatory effects of T propionate (TP) on erection in castrated rats [23,28]. Administration of 5α-DHT with or without the 5α-RIs, 17βTC, restored sexual behavior in long-term castrated male rats and mice suggesting a critical role for 5α-DHT in erectile physiology [29]. Further animal studies showed castration resulted in 50% reduction in erectile response, which was reversed by T treatment [30]. However, treatment with T together with finasteride did not restore the erectile response in castrated animals, suggesting that 5α-DHT is an important hormone in erectile physiology. Administration of 5α-DHT together with finasteride, however, restored nitric oxide synthase (nNOS) expression and activity and also restored the erectile response to electric field stimulation, confirming the role of this metabolite in erectile physiology [30]. In castrated and adrenalectomized rats, treatment with 5α-DHT for 7 days restored erectile function to levels similar to that of control animals [31]. Other studies demonstrated that 5α-DHT treatment in castrated rats improved the erectile response to electrical field stimulation (EFS) [32,33]. Castration in male rats eliminates noncontact erections and this response was restored by 5α-DHT implantation [34,35]. Noncontact erections in animals are thought to be similar to human psychogenic erections [36]. Reduced erectile response and reflex erections were observed in castrated animals [37] and treatment of castrated animals with T or 5α-DHT restored the number of erectile responses and reflex erections. However, only 5α-DHT restored erectile responses and reflex erections, when animals were treated with daily injections of the 5α-R inhibitor MK-434 (1 mg/kg) together with T or 5α-DHT [37]. These findings suggest that 5α-DHT plays a key physiological role in erectile physiology in the animal model and 5α-RIs treatment are likely to produce sexual adverse effect on the erectile response.
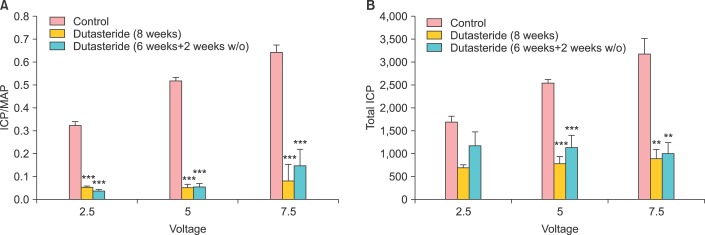
In more recent studies by Oztekin et al. [38] and Pinsky et al. [39] it was demonstrated that treatment of mature animals with dutasteride resulted in reduced serum DHT levels by ~86.5% after 30 days. The intracavernosal pressure (ICP) decreased significantly in animals treated with dutasteride. Similarly, EFS-induced and acetylcholine-induced penile smooth muscle relaxations were also significantly attenuated. More profoundly, connective tissue deposition was markedly increased in the corpus cavernosum of the dutasteride-treated animals. Concomitant with these changes are the markedly reduced expression of neuronal nNOS and increased expression of the inducible NOS, suggesting that dutasteride induces altered gene expression in the corpus cavernosum. Oztekin et al. [38] investigated the effects of dutasteride on erectile function after 6 weeks of treatment followed by a washout period of 2 weeks. The ratio of ICP over mean arterial pressure [MAP] and total ICP in the dutasteride treated animals were significantly decreased when compared with control animals (Fig. 2). Even after two-week washout period the effects persisted and the ICP/MAP was significantly reduced in the dutasteride treated animal, suggesting a persistent effect of the drug on erectile physiology (Fig. 2). Also the endothelium-dependent smooth muscle relaxations were also diminished in the dutasteride treated animals. The authors suggested that discontinuation of dutasteride did not restore erectile function, indicating a time-dependent detriment of dutasteride on erectile physiology (Fig. 2).
A recent study by Zhang et al. [40] reported that treatment of male mature animals for 16 weeks with a daily oral dose of 4.5 mg/kg finasteride significantly reduced 5α-DHT levels and attenuated penile erectile response to EFS of the cavernous nerve. This treatment also reduced the trabecular smooth muscle content and increased connective tissue deposition, reminiscent of the data reported by Traish et al. [41], in the castrated animal model. In addition, finasteride treatment reduced endothelial nitric oxide synthase expression. More importantly, this treatment decreased autophagy and produced deterioration in the ultrastructure of the corpus cavernosum, including mitochondria injury, and increased trabecular smooth muscle cell death (Fig. 3) [40]. These findings, together with the previously reported data from pre-clinical studies point out to serious adverse effects of finasteride and dutasteride on the anatomy, physiology and cell biology of erectile function (Table 1) and suggest that the effects of these drugs may be persistent or irreversible.
2) Evidence from clinical studies
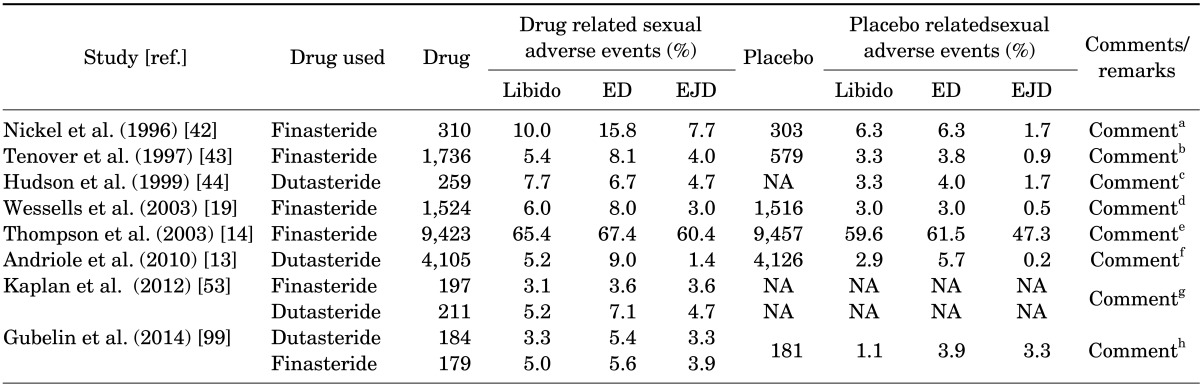
Nickel et al. [42], evaluated the efficacy and safety of 2 years treatment of moderate BPH with finasteride. In the safety analysis, the incidence of adverse events related to sexual dysfunction were significantly higher in the finasteride group than in the placebo group (ejaculation disorder, 7.7% vs. 1.7%; erectile dysfunction (ED), 15.8% vs. 6.3%, respectively; p<0.01 or p=0.01 for both parameters). Tenover et al. [43] also showed that the overall sexual adverse events were greater with finasteride (12.3%) compared with placebo (6%). Finasteride increased the incidence of ED by 7.4% compared to 3.3% in the placebo arm. Similarly, finasteride resulted in loss of libido (4.9%) compared to 2.9% in the placebo arm. In a follow-up to the phase III North American Study, Hudson et al. [44] reported that all patients who completed a 12-month period with 5-mg finasteride were invited for an open-label extension of 4 years. The open-label results showed that ejaculation disorder for years 2, 3, 4 and 5, respectively, were 3.2%, 2.2%, 1.9%, 2.7%, respectively; ED 10.4%, 6.7%, 7.3%, 9.6%, respectively, and decreased libido was 4.8%, 2.7%, 3.9%, 3.7%, respectively. The AUA BPH guideline committee meta-analysis, reported that sexual adverse events due to finasteride and placebo were as follows: libido, 5%, 3%; erectile problems, 8%, 4%; and ejaculation, 4%, 1%, respectively. The PLESS trial [45] enrolled 3,040 men with BPH. Overall maintenance of erection diminished in both groups, but was greater in the finasteride treatment group. In an observational cohort of 14,772 men taking finasteride, ED was the most common adverse event leading to withdrawal from the trial [46]. The AUA clinical practice guideline reported erectile problems in 8% and 4% of patients taking finasteride and placebo, respectively [47].
Edwards and Moore [48] reviewed several clinical trials, three of these trials contained active controls, and 19 trials with placebo groups and reported significantly more men with sexual dysfunction (14%) with finasteride than with placebo at 12 months of treatment (Reduced libido, 5%; ED, 8%; ejaculation disorder, 2%). Incidence rates of sexual dysfunction ranged between 2%-14% with finasteride and 0.6%-7% with placebo. The data from the REDUCE trial [13] also reported that dutasteride resulted in loss of libido/decreased libido of 5.2% and ED of 9% compared to 2.9% and 5.7% in the placebo arm. Similarly in the PCPT trial [14] the incidence of loss of libido (5.8%), ED (5.9%) and ejaculatory disorders (13.1%) higher than that observed in the placebo arm. Similar data was reported by Wessells et al. [19]. The authors suggested that in men who discontinued treatment with finasteride approximately 50% of the adverse sexual events were resolved. However, the authors do not address what happened to the remaining 50% whose adverse sexual events were not resolved after drug discontinuation nor do they address the loss to follow up. Gur et al. [10] in a recent review suggested that decreased circulating 5α-DHT resulting from 5α-RIs use is associated with diminished sexual desire and/or orgasm. The authors suggested that the data from clinical trials with 5α-RIs report prevalence rates of erectile dysfunction of 5%-9%. They further noted that the adverse sexual effects are associated with decreased self-esteem, quality of life, and ability to maintain an intimate relationship.
ED was consistently noted in observational studies as well as in double-blind, randomized, placebo controlled trials (Table 2). Roehrborn et al. [49] and Siami et al. [50] reported that approximately six percent of the patients reported ED in a 2-year follow-up to the CombAT trial [49,50]. Hudson et al. [44] reported that ED occurred in 6.7% and 4.0% of patients treated with dutasteride or placebo, respectively, in a trial for treatment of BPH [44]. Desgrandchamps et al. [51] reported 7% of the drug related adverse effects were ED. In the PROSPECT study, ED was established but determined subjectively in an open-ended interview [52].
In a retrospective analysis of data from consecutive patients treated at a single clinic, Kaplan et al. [53] showed that both finasteride and dutasteride resulted in sexual adverse effects. Approximately 42.6% discontinued treatment with finasteride and 57.5% discontinued treatment with dutasteride and 29.4% of the patients were lost to follow up in the finasteride group and 36.5 lost to follow up in the dutasteride group, respectively. ED was reported in 3.6% in the finasteride and 7.1% in the dutasteride, respectively. Ejaculatory dysfunction was reported in 3.6% in the finasteride group and 4.7% in the dutasteride group respectively. Decreased libido was approximately 3.1% in the finasteride and 5.2% in the dutasteride group, respectively. The authors suggested that dutasteride elicits more sexual side effects and breast complications than finasteride [53]. Chi and Kim [54] investigated the effects of dutasteride treatment during a 1-year follow-up period in Korean men. The erectile function domain (EF) of the International Index of Erectile Function decreased significantly after 1 month and remained significantly reduced even after 12 months of treatment. Orgasmic function and sexual desire were significantly reduced but slowly recovered after six months. The authors concluded that "After 1 month of treatment, dutasteride therapy resulted in a significant reduction in all investigated sexual functions. Overall, recovery in sexual function was noted at 3 months, and orgasmic function and sexual desire were restored to baseline levels at 6 months. However, EF was still significantly reduced at 12 months". In a recent study Fwu et al. [55] investigated the effects of finasteride with or without combined therapy with ABs on sexual function and found that men assigned to finasteride or finasteride combined with doxazosin experienced a worsening of several domains of sexual function compared to placebo [55].
To this end, the data from a number of clinical studies (Table 2) clearly showed that in some patients, treatment with 5α-RIs diminished libido, erectile, and ejaculatory function. These adverse effects may not be significant in the realm of the overall studies, but for the individual patient this is a serious loss of quality of life and should be given serious considerations prior to commencing therapies with these drugs. 5α-RIs' therapy, while improves urinary symptoms in patients with BPH and may prevent hair loss in patients with MPHL, this therapy produces significant adverse effects in some individuals including loss of libido, ED, ejaculatory dysfunction, and potential depression. These adverse events are serious enough to preclude them from pursuing such therapy. The effects of these agents on vascular health should also be noted in light of recent findings that patients treated with 5α-RIs therapy had significant adverse cardiovascular events [13].
2. 5α-RIs therapy contributes to high Gleason grade PCa
The premise put forth by Huggins (1941) suggested that PCa is androgen-dependent and androgen deprivation results in tumor shrinkage [56]. Androgen-dependency of PCa is predictable because tumor cells are derived from normal prostate cells, which retain expression of the androgen receptor (AR). Androgen deprivation therapy (ADT) of PCa initially results in tumor regression, but the cancer often relapses into androgen-independent cancer within 18-24 months and tumors no longer require androgens for growth [5,6].
Because conversion of T to 5α-DHT is a critical pathway in normal prostate growth and function, inhibition of 5α-R activity in the prostate results in a reduction of intraprostatic 5α-DHT levels and a decrease in prostate growth and size [57]. There is no evidence that T or 5α-DHT causes initiation, promotion or development of PCa [5,6,58,59]. This hypothesis was further supported by the recent study of Muller et al. [59] in which patients enrolled in the Placebo arm of the REDUCE trial [13] showed no association between baseline total T nor 5α-DHT and PCa. The authors concluded that "baseline serum T and 5α-DHT levels were unrelated to prostate cancer detection or grade" [13,59]. This raises the question-how could 5α-RIs be useful as chemo-preventative agents if PCa development is unrelated to T or 5α-DHT? As stated by Walsh (2010) in the FDA panel discussion in 2010, "No clinical benefit has been demonstrated in patients with prostate cancer treated with PROSCAR" [16]. In addition, the potential reduction in the incidence of PCa in the general population with the use of these agents may not exceed 10%, which is not statistically significant [16]. Furthermore, the documented increase in Gleason high-grade PCa tumors in response to treatment with these agents proves that these drugs do not prevent development and growth of PCa; they merely prevent biopsies due to reduction in prostate volume. Most importantly, if 5α-RIs indeed prevent PCa development, then how come they are not approved for treatment of PCa?
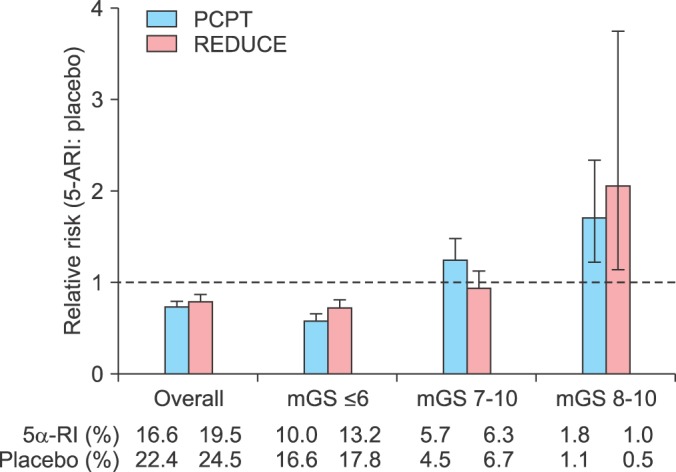
As suggested by Crawford et al. [60], Justman [61], and Walsh [62,63] four important points need to be considered before the decision is made to use 5α-RIs for chemo-prevention of PCa. These are: (1) 5α-RIs do not prevent PCa development and therefore, these drugs have no significant effect on reducing the real risk of metastatic PCa in men; (2) 5α-RIs reduce the risk of detecting low-grade tumors, but do not increase survival or reduced mortality [17]; (3) if these drugs indeed chemo-prevent PCa, then why aren't they used in the treatment of PCa?; and (4) to reduce the overall risk of PCa in the population, 5α-RIs as chemo-preventative agents must be entirely free of any side effects [60,61,62,63]. Theoret et al. [15] reported that even after re-evaluation of the data from the PCPT [14] and the REDUCE trial [13] with the revised Gleason scoring system, these drugs were shown to increase the incidence of Gleason high-grade PCa tumors. The authors concluded that "the trade-off inherent in using a 5αRI for prostate cancer prevention is the acceptance of one additional high-grade cancer in order to avert three to four potentially clinically relevant low-grade cancers" [15]. Put more simply, when using 5α-RIs as chemo-preventive agents - the risks outweigh the benefits.
This is not surprising since the recent follow-up data on the PCPT have clearly shown that there are no significant benefits in terms of reducing mortality and increasing survival [17]. As Ehdaie and Touijer [64] pointed out, while the medical community is eager to provide early intervention for patients with high prostate specific antigen levels after therapy, it remains critical to weigh the harm of treatments against the uncertain benefits to the patients. It is incumbent that physicians critically evaluate the current evidence and weigh the risks and benefits of such interventions [64]. Since the PCPT [14] and REDUCE trials [13], used PCa incidence as an end point instead of mortality, there is little that one can conclude about the chemo-preventive nature of these agents and their ability to prolong survival and improve quality of life [65].
Data from a large, long-term Randomized Control Trial (RCT ARI40005) raised a serious concern regarding the safety of 5α-RIs, dutasteride, because of significant increase in the risk of "cardiac failure" [13,66,67]. As pointed out by Justman [61] some of the unidentified risks of 5α-RIs may be attributed to the limitations of the clinical trials. For example, these trials had no primary or secondary endpoints to detect cardiovascular events; therefore, some of the events may have been overlooked or underestimated. Moreover, since cardiovascular events were not investigated as part of the primary or secondary endpoint, clinical trial investigators may have had different interpretations and may have not given them serious consideration [16,61,67].
Several studies have attempted to evaluate the efficacy of 5α-RIs in reducing tumor growth in the animal models. Interestingly, the study by Umekita et al. [68] showed that various PCa tumor cell lines respond differently to androgen treatment. For instance, the tumor cell line, LNCaP 104-R2, when implanted in castrated, nude, male mice continued to grow. However, treatment with TP resulted in tumor regression not tumor growth. Remarkably, treatment with TP together with finasteride resulted in increased tumor growth. These findings suggest that ADT may not necessarily be beneficial for treatment of all forms of PCa and that androgen replacement therapy may be a potential treatment for some forms but not all forms of PCa [68]. Recently, several small clinical trials have reported using testosterone therapy in patients with localized PCa, after radical prostatectomy or brachytherapy. These studies found positive results based on lack of biochemical recurrence in the treatment arms [69].
Since PCa is a heterogeneous disease, different tumors possess different metabolic properties. In a study using androgen-insensitive PCa cell lines in an animal model, it was reported that the 5α-RIs, finasteride and dutasteride, did not inhibit tumor incidence or tumor growth suggesting the limited potential benefits of 5α-RIs as chemo-preventive agents in all forms of PCa.
It should be noted that 5α-RIs alter cellular biology with uncertain outcomes. For example, treatment of animals with 5α-RIs resulted in marked increase in the expression of the AR [70]. The implication of such dysfunctional metabolism may contribute to loss of androgen dependence and to acquisition of high-grade tumors. In addition, Yun et al. [71] postulated that finasteride increases expression of hemoxygenase-1 and other related factors in PCa cell lines (PC-3). The authors suggested that finasteride-induced alteration in gene expression may be in part responsible for finasteride-induced high grade prostate tumors [71]. It is not surprising that Theoret et al. [15] reported a significantly number of high grade tumors (Fig. 4), with finasteride and dutasteride in the data from the PCPT [14] and REDUCE trials [13] respectively, even after using the revised Gleason scoring system [15].
The overwhelmingly negative decision by the FDA panel of scientists and medical experts [16] speaks volumes and raises a significant concern about the safety and the efficacy of these drugs as chemo-preventive agents for PCa, given that they elicit many serious adverse side effects [16]. With the postmarketing increase in reported adverse events, the FDA has mandated a revision to the labeling for all 5α-RIs [16]. The labeling changes included a warning of increased sexual dysfunction, depression, and increased risk of high-grade PCa.
3. Potential adverse effects of 5α-R therapy on the CNS
The CNS has the capacity and the machinery to synthesize a host of neurosteroids [72]. Neurosteroids play a pivotal role in modulating neural activity through interaction with neurotransmitter receptors and neurotransmitter-gaited ion-channels [73]. These neurosteroids interact with a host of neurotransmitter receptors and modulate seizure susceptibility, anxiety, stress, and depression [74]. 5α-R reaction is the rate limiting step in the conversion of testosterone, progesterone, cortisol, corticosterone, and DOC into their respective 5α-dihydro-deratitves, which serve as precursors for 3α-hydroxysteroid dehydrogenase which transfroms such precursors into their respective neurosteroids (androstanediol, allopregnanolone [AP], tetrahydrocortisol, tetrahdyrocorticosterone, and tetrahydrodeoxycorticosterone) (Fig. 1) [1,2]. All three isoforms of 5α-R are expressed in the various regions of brain and are thought to be critical for brain development since fetal brain express high concentrations of 5α-R [1,2].
It has recently been shown that patients who had been treated with finasteride have reduced or undetectable levels of neuroactive steroids in their cerebro-spinal fluid and plasma, and exhibited higher levels of precursor steroids [75]. This observation strongly suggests that 5α-RIs have a deleterious effect on the biosynthesis and function of neurosteroids in the central nervous system. Finasteride treatment resulted in decreased levels of 5α-DHT and 3α, 5α-tetrahydroprogesterone (AP) and increased levels of testosterone supporting the hypothesis that deleterious effects of finasteride may be persistent or irreversible. This may explain some of the noted symptoms such as anxiety, depression and suicide in patients who have been treated with finasteride [76].
NEUROPROTECTIVE EFFECTS OF NEUROSTEROIDS
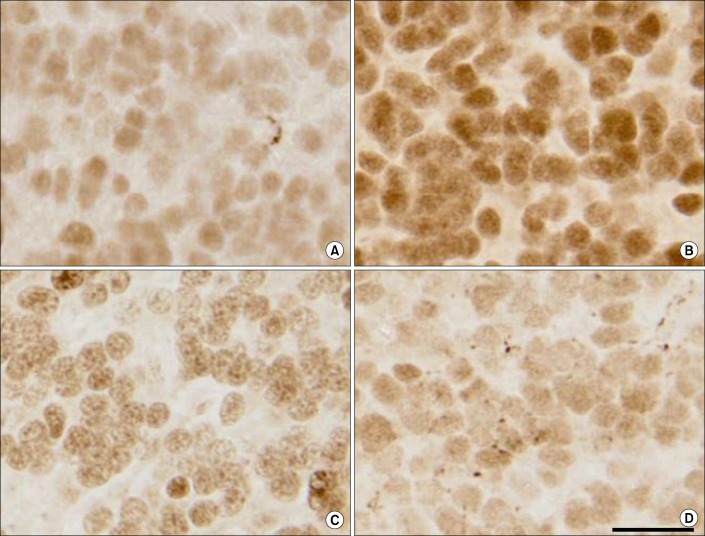
Neuroactive steroids elicit important neuroprotective effects during trauma and injury to the central nervous system [75]. AP is shown to be beneficial in the treatment of traumatic brain injury, attenuating edema, trauma, stress, inflammation, apoptosis, and reducing oxidative stress [76,77,78]. AP is not only a protective agent in ischemia, but also in maintaining blood brain barrier integrity, and in memory and learning [78,79,80,81]. Studies on CNS injury in which asphyxiation was induced in fetal sheep to stimulate neurological stressors, in the presence or absence of finasteride, showed an increase in apoptotic cell death in the cerebellum and hippocampus in the animals treated with finasteride (Fig. 5) [82]. Furthermore, treatment with an AP analogue, alfaxalone, prevented cell death as assessed by the increase in activity of caspace-3 and expression of ki-67 protein. This observation suggests that AP exerts a neuroprotective role in the brain, which is inhibited by finasteride.
The neuroprotective effect of AP was further illustrated during injury to the rat hippocampus slices induced by tributyltin treatment, which resulted in significant cell death. Administration of progesterone (P) with finasteride showed similar cell death to that induced by tributyltin treatment, in the various regions of the hippocampus. In contrast, P treatment without finasteride provided a protective effect. This is attributed to the conversion of P to 5α-DHP by 5α-Rs and to AP by 3α-HSD. To confirm that this is due to the neuroprotective effect of AP, the latter was administered with or without finasteride. While the tributyltin induced cell death was significant, administration of AP with and without finasteride produced a markedly protective effect as assessed by the reduced cell death [83]. These findings suggest that 5α-Rs play a pivotal role in neuroprotection.
Neurosteroid synthesis in the hippocampus is suggested to be critical for neuroplasticity in the brain [84]. Inhibition of 5α-R by finasteride is thought to contribute to reduced neuroplasticity due to structural changes resulting from inhibition of neurogenesis in the hippocampus. Finasteride treatment in mice showed decreased cell proliferation in the hippocampus, suggesting that inhibitors of 5α-R blocks neurogenesis [84].
As reported by Mellon [85] in an adult animal model of Niemann-Pick disease type C the biosynthesis of AP is significantly reduced, and this was supported by significant reduction in the activity of 5α-R and 3α-HSD in the brain of these animals [85]. The activity of 5α-R was markedly reduced with the progression of the disease. This reduced 5α-R activity may contribute to the observed accumulation of cholesterol in neurons and gangliosides, cerebro, perkinje cell degeneration, dysmyelination and neurodegeneration [85]. Thus, loss of 5α-R and 3α-HSD activity is attributed to loss of neurons expressing these enzymes. Treatment with AP early at postanatal day 7 was found most effective in preventing neurodegeneration and correlates with reduced tremor, ataxia, and increased lifespan. In the animal model, these findings indicate that the loss of neurosteroid biosynthesis may be responsible for the disease state and its progression. Therefore inhibition of 5α-R by finasteride and dutasteride in the course of treatment of nonlife threatening conditions, such as male pattern baldness (alopecia) or BPH may have detrimental effects on the CNS.
It has been reported that AP levels were significantly decreased in postmortem human brains of Alzheimer disease (AD) patients [86]. An inverse correlation was noted between AP levels and the degree of neurological degeneration in pathological section of AD patient [86]. One of the interesting findings was that pregnenolone levels were greater in the temporal cortex of AD patients suggesting that this may be a compensating mechanism for reduced 5α-R activity. We speculate that 5α-RIs may contribute to reduced levels of neurosteroids in the CNS and this may enhance the progression of neurodegenerative disease, such as AD.
Conversion of polyprenol into dolichol is critical for N-glycosylation of membrane proteins [3]. 5α-R type 3 was shown to be critical for catalyzing this reaction [3]. A mutation in this enzyme lead to mental retardation and opthamologic and cerebral defects [3]. Intraoperative Floppy Iris Syndrome (IFIS) is also a complication experienced during cataract extraction. The syndrome is defined by floppy, or flaccid iris. Studies have indicated a correlation between finasteride and the occurrence of cataracts and IFIS indicating inhibition of glycosylation may contribute to this pathology [87].
EPILEPSY/CONVULSION
P is an effective anticonvulsing agent in humans [88]. The anticonvulsive properties of P diminished when animals were treated with finasteride. In a mouse model of pentylenetetrazol-induced seizures, there was an approximately 50% decrease in the protective effect of P in mice when treated with finasteride [89]. Higher dosages of finasteride produced more persistent symptoms of pentylenetetrazol-induced seizures [89]. However, when AP was administered together with finasteride, the pentylenetetrazol seizure activity was reversed. This finding indicates that the antiseizure properties of P are attributed to its metabolite AP emphasizing the critical role of 5α-Rs [89,90,91].
DEPRESSION
Anxiety is often found as a comorbidity of depression. The administration of AP produces antidepressant and anxiolytic effects [92,93]. Coadministration of finasteride and P blocked P's anxiolytic effect [94]. This finding suggests that a metabolite of progesterone is responsible for the anxiety reducing effect of P. An inverse relationship between levels of AP and depression has been demonstrated in male patients with depression [95]. Preclinical studies have suggested that reduction in AP levels by 5α-RIs may contribute to depressive symptoms [96]. Increased depressive symptoms are thought to be linked to finasteride treatment [97]. A statistically significant correlation was observed between use of finasteride and depressive symptoms [98]. Persistent side effects have been noted even after discontinuation of finasteride treatment [76] from 3 months to 11 years, suggesting that the adverse effects of finasteride may be permanent [97].
SUMMARY
We wish to point out from the outset that 5α-RI therapy was FDA-approved for treatment for LUTS related to BPH. Treatment of symptomatic men with finasteride or dutasteride results in approximately 17%-19% reduction in prostate volume, thus alleviating urinary tract symptoms [8,13,14,53]. Furthermore, 5α-RI therapy reduces urinary tract infection and minimizes the risk of potential acute urinary retention and the need for surgical intervention related to BPH. These positive effects of finasteride and dutasteride contribute to improvement in the quality of life in men suffering from BPH symptoms [8,53]. However, a substantial body of evidence exists which points to serious and potentially ill-health effects of 5α-RIs' therapy. These include loss or reduced libido, erectile dysfunction, orgasmic and ejaculatory dysfunction (Table 2) [99], development of high grade PCa tumors (Fig. 4), potential negative cardiovascular events, and depression. The side effects are potentially harmful in some individuals and in young men may be persistent or irreversible [100]. The argument that the benefits of these drugs outweighs the risks is slowly eroding in the face of new emerging scientific evidence from preclinical (Figs. 2, 3; Table 1) and clinical studies (Table 2). The available data demonstrate that such drugs do pose serious adverse effects, especially in a subset of men who may have the predisposition to be affected severely. This is also corroborated by the overwhelming FDA disapproval of these drugs for the chemoprevention of PCa. The FDA mandated relabeling issued in 2011 equally points to the dark side of these drugs on human health. Physicians need to be aware of the adverse side effects of these drugs and are encouraged to share this information with their patients prior to commencing therapy with finasteride or dutasteride.

 XML Download
XML Download