

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Translocation renal cell carcinoma (RCC), also known as juvenile RCC, is a new entity that was recently added to the 2004 World Health Organization RCC classification. These tumors are characterized by different chromosome translocations resulting in gene fusion between Xp11.2 and different transcription factor genes. Most translocation RCCs (about 90%) involve the transcription factor E3 (TFE3). Another less common translocation, t(6;11) (p21;q12), involving fusion between the Alpha gene and the transcription factor EB (TFEB) has also been reported.
In the literature, most case reports and series of translocation RCC concern pediatric and young adult populations from Western countries [1,2]. In the French West Indies, the population is mainly issued from Sub-Saharan African countries and the prevalence of sickle cell disease is 0.35% of births. Usually, renal tumors of sickle cell disease are represented by renal medullary carcinoma (RMC) [3]. Here we present the first case of a young woman from French West Indies with sickle cell anemia who developed a translocation RCC t(6;11)(p21;q12). In the present case study, a literature review of translocation RCC and links with sickle cell disease and RMC are presented and discussed.
CASE REPORT
A 27-year-old woman from the French West Indies who was followed at Pointe-a-Pitre University Hospital for a sickle cell anemia was referred for a left renal tumor. She had a personal history of cholecystectomy and acute thoracic syndrome.
A left renal mass issuing from the renal sinus was discovered incidentally on an abdominal ultrasound performed for the patient's annual sickle cell anemia checkup. The clinical examination revealed a weight loss of 3 kilograms. There was no hematuria, no flank pain, no asthenia, and no lumbar contact (ECOG=0).
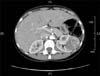
A computed tomography (CT) scan confirmed a left renal tumor of 34 mm×86 mm, which seemed to develop from the renal medulla, that was heterogeneously enhanced with contrast agent (Fig. 1) and several perihilar adenopathy (the largest being 17 mm). Thoracic CT scan revealed no metastasis.
A left laparoscopic radical nephrectomy was performed after specific preoperative care for sickle cell disease: exchange transfusion for hemoglobin S <40%, hyperhydration, chest physiotherapy, and prevention against acidosis and hypothermia. There were no postoperative complications.
Final pathological examination showed a 60-mm well-defined medio-renal tumor with a brownish-red appearance. The 11 removed lymph nodes were negative. Microscopically, the tumor showed biphasic growth patterns consisting of nests of eosinophilic and clear cells (Fig. 2). Immunohistochemical analysis was performed, using a panel of antibodies including CD10, RCC marker, vimentin, α-methylacyl-coenzyme A racemase (p504s), human melanoma black 45 (HMB45), microphthalmia transcription factor (MiTF), TFE3, and TFEB. The tumor cells revealed diffuse nuclear TFEB staining, demonstrating a translocation RCC t(6;11)(p21;q12). They were also positive for vimentin, p504s, and HMB45 but in less than 10% of tumor cells. This tumor was classified as stage pT1bN0M0 Fuhrman grade 2.
Twelve months after the surgery, the patient was in excellent condition and no recurrence was noticed on the CT scan.
DISCUSSION
To our knowledge, this report is the first case described in the literature of a translocation RCC in a patient presenting with sickle cell anemia. Indeed, people with the sickle cell condition usually develop RMC, a variety of high-grade RCC described by Davis Jr et al. [3] for the first time in 1995.
Classically, RMC affects children and young adults, with a mean age of 19 years (ages range from 5 to 69 years) and with a male-to-female ratio of 2:1 [4]. In the literature, all patients are black and the majority have sickle cell disease [4]. RMC is designated as the seventh sickle cell nephropathy (the others being macroscopic hematuria, papillary necrosis, nephrotic syndrome, renal infarction, pyelonephritis, and inability to concentrate urine) [3].
RMC is an aggressive tumor with a poor prognosis; 90% of the patients are usually diagnosed at a metastatic stage [4] with a median survival of 15 to 18 weeks after nephrectomy [3-6]. Since 1995, 160 cases have been reported in the literature, and only one of them had a homozygous pattern [4]. The relationship between RMC and sickle cell anemia can be explained by chronic hypoxia in the renal medulla due to vaso-occlusion. The chronic hypoxia stimulates the expression of growth factors such as hypoxia inducible factor and vascular endothelial growth factor, which are well known in the neoangiogenesis of renal tumors [6,7].
In this report, before the surgery the patient was thought to have an RMC and we did not expect the pathological report conclusion of a translocation RCC. Translocation RCC represent one third of renal carcinomas in young patients [8]. They are mainly described in children and young adults from Western countries. The average age at diagnosis is around 24 years with a female-to-male ratio of 2.5:1. Patients usually present symptomatically with hematuria, abdominal pain, or an abdominal mass, whereas one third remain asymptomatic [1,4,9].
RCCs are usually large (an average of 6-7 cm in diameter) and their macroscopic appearances are similar to clear cell carcinoma: yellowish to brownish, well circumscribed, and sometimes associated with necrosis or hemorrhage. Microscopically, they have a papillary or solid architecture, with a mixture of clear and eosinophilic cells. More specifically, the microscopic appearance of translocation RCC t(6;11)(p21;q12) as described in our patient has a two-fold component involving nests of large eosinophilic cells and smaller epithelial cells clustered around hyaline nodules [8].
Diagnostic confirmation of translocation RCC t(6;11) (p21;q12) requires immunohistochemical identification of the nuclear transcription factor (TFEB) and/or cytogenetics and/or molecular biology (fluorescence in situ hybridization, polymerase chain reaction) identification of the translocation [4,8]. Most carcinomas with translocation (90%) involve TFE3 located on chromosome Xp11.2. The most common gene fusions are ASPL-TFE3 and PRCC-TFE3. Another less common group of RCC with translocation was described in 2001: the translocation t(6;11)(p21;q12), which results in fusion of the Alpha gene, located on chromosome 11q12 with the TFEB gene on chromosome 6p21 [1,8]. Carcinoma with translocation involving the TFEB and TFE3 belong to the family of RCC with MiT translocation involving MiTF and transcription factor EC, which were grouped by Argani and Ladanyi [9] within the same family of translocation RCC MiTF/TFE.
These translocation RCCs seem to have a relatively indolent evolution, despite a common diagnosis at an advanced stage with lymph node metastasis [1,2].
The optimal treatment for translocation RCC remains to be determined. It is thought that for localized tumors, surgery is adequate and that lymph node involvement is not an adverse prognostic factor.
A series including 31 patients with translocation RCCs reported only 2 translocation RCC t(6;11)(p21;q12) [2], but no data are available about the specific evolution of those TFEB translocation RCCs.
The association between "MiTF/TFE family translocation carcinomas" and sickle cell anemia, which is described here for the first time, might be coincidental or might be related to genetic abnormalities of sickle cell anemia and translocation RCC t(6;11)(p21;q12), both of which are located on chromosome 11. This relation remains unclear.

 XML Download
XML Download