

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Erectile function is a complex neurovascular process that requires the integration of a host of signaling pathways, leading to increased blood inflow to the corpora cavernosa and reduced blood outflow permitting entrapment of blood under pressure and engorgement of the penis [1-6]. Upon sexual stimulation, the release of neurotransmitters from the cavernosal nerves brings about vascular and trabecular smooth muscle relaxation. This permits increased blood flow from the cavernosal arteries and activation of endothelial cells by producing nitric oxide (NO) in response to shear stress. The relaxation of the vascular smooth muscle of the cavernosal artery, helicine arterioles, and the trabeculae increases intracavernosal pressure as the result of the accumulation of blood under pressure [1-6]. Vasodilation of the arterioles that feed the lacunar spaces and relaxation of corporeal smooth muscle, which surrounds these spaces, contributes to increased intracavernosal pressure and engorgement of the corporal bodies as a result of filling with blood, which results in expansion of the corpora bodies against the tunica albuginea. The accumulation of blood under pressure compresses the subtunical venules and impedes blood outflow, resulting in the entrapment of blood under pressure and a concomitant increase in the intracavernosal pressure, veno-occlusion, and subsequent erection.
The veno-occlusion mechanism is regulated by local release of 1) adrenergic and cholinergic neurotransmitters; 2) nonadrenergic, noncholinergic neurotransmitters; and 3) vasoactive agents produced by the vascular endothelium [3-5]. The sympathetic nervous system releases norepinephrine, which acts through the α-adrenergic receptor pathway, thus modulating the contractility of vascular and trabecular smooth muscles, leading to detumescence. Nonadrenergic, noncholinergic neurotransmitters (e.g., neural nitric oxide, vasoactive intestinal polypeptide) released from the parasympathetic nerves facilitate vascular and trabecular smooth muscle relaxation. In addition, NO and vasoactive substances, such as prostacyclins and prostaglandins, released by the vascular endothelium also stimulate smooth muscle relaxation, producing increased intracavernosal pressure, veno-occlusion, and erection [1-5,7]. Alterations or interferences in the mechanisms regulating vascular and trabecular smooth muscle contractility contribute to veno-occlusive dysfunction and ultimately erectile dysfunction (ED) [1,4,5].
Androgens play an important role in erectile physiology. It is believed that androgens are critical not only for the growth and function of the trabecular smooth muscle, but also in maintenance of the function of cavernosal and dorsal nerves as well as the function of the vascular endothelium. In this review, we focus mainly on the role of androgens in modulating endothelial function and in the development, differentiation, maturation, migration, and homing of endothelial progenitor cells (EPCs) and their role in maintaining erectile physiology.
ROLE OF THE ENDOTHELIUM IN ERECTILE PHYSIOLOGY
The vascular endothelium plays a critical role in the vasculature [2,7-11]. The endothelium serves as a barrier between circulating blood and blood-borne elements and the surrounding tissues [8,12]. The endothelium is critical for angiogenesis and vascular repair. Furthermore, the vascular endothelium produces a host of regulatory factors, including NO, prostaglandins, endothelins (E-1, E-2, E-3), prostacyclin (PGI2), and angiotensin II, among others [8,13,14]. These signaling molecules regulate smooth muscle contractility and subsequent vasoconstriction and vasodilation and ultimately regulate blood flow to the penis. In erectile tissue, the signaling molecules produced by the endothelium regulate the contractility of the trabeculae and are deemed critical in normal erectile physiology [14]. Thus, the endothelium regulates vascular tone and erectile physiology via autocrine, paracrine, and endocrine mechanisms [7,13,14].
In erectile tissue, the endothelium lining the vascular bed and the trabeculae in the penis plays a critical role in erectile function. The increased blood flow through the cavernosal artery increases shear stress, which causes the endothelium to synthesize and release NO. NO relaxes the vascular smooth muscle and increases blood flow to the corpora cavernosa. The endothelium lining the lacunar spaces responds to this increased blood flow with increased synthesis and release of NO, which facilitates the relaxation of the trabecular smooth muscle of the corpora cavernosa. These signaling mechanisms originating from the endothelium are considered critical for the regulation of penile physiology and erections.
The endothelium responds rapidly to signaling by a variety of stimuli and is instrumental in the recruitment of immune and blood cells to sites of endothelial injury [1,7,8,14-17]. The endothelium regulates vascular homeostasis by activating platelets and inflammatory and immunological responses as well as by controlling vascular permeability, smooth muscle cell proliferation, and angiogenesis [1,7,14,18-20]. Thus, the endothelium plays an important role in the repair of injury to the vascular bed by replacing injured endothelial cells with mature EPCs. Baumhakel [21] investigated the relationship between circulating EPCs and erectile function by using CD133+ as a marker to identify the EPCs. Patients with ED had significantly lower levels of circulating CD133+ cells than did patients with normal erectile function. Circulating CD133+ cells showed an inverse correlation with ED, and increasing the number of these circulating EPCs resulted in a decreased likelihood of ED [21].
Endothelial dysfunction is associated with reduced endothelial nitric oxide synthase (eNOS) expression; increased production of the asymmetric dimethylarginine (ADMA); increased production of reactive oxygen species (ROS); increased synthesis and release of ET-1, E-2, and E-3; increased production of inflammatory cytokines such as tumor necrosis factor-α (TNF-α); increased expression of markers of cell adhesion such as E-selectin, intracellular adhesion molecule (ICAM), and vascular cell adhesion molecule (VCAM); deregulation of fibrinolytic factors such as von Willebrand factor (vWF); inability to regenerate endothelium from EPCs; increased endothelial cell apoptosis; increased cellular and vascular permeability; and increased vascular tone [22].
RISK FACTORS CONTRIBUTING TO ENDOTHELIAL DYSFUNCTION AND POTENTIAL REPAIR MECHANISMS
A common link underlying the pathophysiology of cardiovascular disease (CVD) and ED is vascular insufficiency attributed to endothelial dysfunction. A host of risk factors, including insulin resistance, type 2 diabetes mellitus, dyslipidemia, hypertension, obesity, metabolic syndrome, oxidative stress, atherosclerosis, smoking, and hyperhomocysteinemia, are thought to contribute significantly to endothelial dysfunction [14,22-27].
EPCs play an important role in vascular repair and angiogenesis and replacement of damaged endothelial cells in blood vessels [13-15,21,28-34]. EPCs were first described as a population of circulating cells able to engraft in areas of physiologic or pathological neovascularization, angiogenesis, and endothelial cell repair by acquiring an endothelial phenotype, expressing vWF, and incorporating Dil-Ac-LDL [8,13,15,28,35-38]. It is believed that endothelial injury is likely to be repaired by mature circulating EPCs.
EPCs are characterized as a population of CD34+ hematopoietic progenitor cells that can differentiate in vitro into an endothelial lineage, expressing a number of cell surface markers similar to those expressed by angioblasts and hematopoietic cells, which suggests a common precursor [15,28,29]. "Putative endothelial progenitor cells" isolated from human peripheral blood express two antigens, CD43 and fetal liver kinase (Flk-1). Peichev et al. [39] identified a CD133+/vascular endothelial growth factor receptor 2+ (VEGFR2+) population that differentiates into endothelial cells in vitro and is distinguishable from mature CD34+/VEGFR+ endothelial cells found on the vessel wall; note that the VEGFR is also known as kinase-inert domain receptor (KDR) [28,31,37,40].
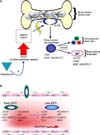
The pathway by which EPCs originate in the bone marrow and a host of biochemical activators, which are thought to play a critical role in the development and differentiation of the precursors of circulating EPCs, are depicted in Fig. 1A. Briefly, the bone marrow produces hemangioblasts, which are thought to be a common precursor of hematopoietic stem cells as well as bone-marrow-derived angioblasts. The angioblasts undergo further differentiation in the presence of the appropriate activators into EPCs [30,31]. Fig. 1B illustrates the differentiation of early EPCs to late EPCs and the expression of various specific markers that are detected on the surface of the mature EPCs during circulation in the bloodstream.
Note that EPCs derived from bone marrow exhibit a wide range of heterogeneity; thus, there is no clear consensus on which specific antigenic profile or surface markers best identify EPCs [41]. However, the three most commonly used antigenic markers for the detection of EPCs are as follows: 1) CD34, an adhesion molecule; 2) CD133, a surface antigen with unknown function that is closely related to immature EPCs; and 3) Flk-1/KDR, or VEGF-2, which indicates early endothelial differentiation [41]. Other markers include AC133, CXCR4, and CD105 (Endoglins). It is believed that mature EPCs can home in on sites of endothelial injury and are involved in the vascular repair processes. Circulating progenitor cells represent a more immature pool of circulating cells with characteristics similar to those exhibited by EPCs. Both progenitor cells and EPCs originate from hematopoietic stem cells of the bone marrow, which then migrate into the peripheral circulation, home in on sites of neovascularization, and differentiate into mature endothelial cells and play a critical role in endothelial repair processes.
Several clinical trials have reported exciting findings regarding the potential use of EPCs in the treatment of various vascular disorders attributed to endothelial dysfunction. In the Transplantation of Progenitor Cells and Regeneration Enhancement in Acute Myocardial Infarction trial (TOPCARE-AMI trial), infusion of ex vivo expanded bone marrow EPCs into patients with a history of myocardial infarction resulted in enhanced myocardial viability and increased ventricular ejection fraction [42]. EPCs have also been used diagnostically as biomarkers of disease detection or progression. For instance, a large number of studies have shown significant changes in the number and function of EPCs in diseases such as diabetes, hypercholesteremia, pulmonary hypertension, rheumatoid arthritis, and chronic renal disease [14,15,21,22,32,33,43, 44]. Also, reduced EPC number and altered proliferation and migration profiles have been correlated with smoking, aging, and chronic inflammation. These findings further support a critical role of EPCs in vascular repair mechanisms and homeostasis.
ANDROGEN MODULATION OF ENDOTHELIAL FUNCTION IN ERECTILE PHYSIOLOGY
In the vasculature, androgens have been shown to modulate endothelial function via genomic and nongenomic mechanisms. In addition, androgens are the precursor for estrogens, which also modulate endothelial function [7,45,46]. Several studies have demonstrated that mature endothelial cells express androgen receptors (ARs), which suggests that androgens elicit a direct genomic-mediated action through the activation of the classic AR [7,47]. Androgens (testosterone and 5α-DHT) also promote nongenomic action via interactions with putative membrane ARs (nontranscriptionally) [7,46]. The results of preclinical and clinical studies have suggested that the relationship of endothelial function in response to androgen is complex and encompasses structural as well as cell signaling pathways. Testosterone deficiency (TD) has been shown to contribute to endothelial damage and dysfunction, and androgen therapy enhances endothelial repair [20,48-52]. Androgens increase the synthesis and release of NO in the vascular endothelium [2,48,53-55]. A recent report has suggested that androgens stimulate EPC proliferation via an AR/VEGF-mediated mechanism [56]. In addition, real-time real time quantitative reverse transcription polymerase chain reaction analysis showed that 5α-DHT induced AR, cyclin A, cyclin D1, and VEGF gene expression in a dose- and time-dependent manner [56]. In in vitro studies using the AR antagonist Casodex and specific AR siRNA, a marked reduction in 5α-DHT-induced endothelial cell proliferation and targeted gene expression was shown, which suggests that these effects are AR-dependent. In addition, inhibition of VEGF receptor signaling or expression produced a dose-dependent blockade of 5α-DHT-induced endothelial cell proliferation and cyclin A gene cell expression. These findings strongly suggest that androgens stimulate endothelial cell proliferation via upregulation of VEGF-A, cyclin A, and cyclin D1. Androgens also promote angiogenesis via cellular mechanisms involving VEGF, a specific cytokine that plays an important role in mitosis in the vasculature. Specifically, binding of VEGF to its receptors on endothelial cells promotes and enhances vascular permeability and facilitates angiogenesis and revascularization [40]. More interestingly, Godoy et al. [57] linked androgens to the upregulation of VEGF and mitotic cyclins, which are known to increase EPC proliferation [57]. In clinical studies it was demonstrated that flow-mediated dilation (FMD) is reduced in patients with TD and is enhanced in response to testosterone therapy [23,58-60].
Lu et al. [50] demonstrated that androgen deprivation by castration or by inhibition of the enzyme 5α-reductase resulted in profound physical and structural changes in endothelial structure in rat aorta as detected by transmission scanning electron microscopy analyses. In aortic tissues from control animals, the endothelium appeared smooth and freely compliant. In contrast, the endothelium in the aorta of castrated animals or intact animals treated with the 5α reductase irreversible inhibitor finasteride were severely crimpled, coarse, and protuberant, and the connections between cells were disrupted and many red blood cells were noted to adhere to the endothelial surface [50]. These findings suggest that not only testosterone but also 5α-DHT is critical for endothelial function. More importantly, the aortic endothelium from castrated animals treated with testosterone undecanoate showed marked structural recovery, suggesting vascular repair [50]. On the basis of these observations, it was suggested that androgen deprivation, via castration or treatment with 5α-reductase inhibitor, produced injuries in aortic endothelial cells [7,50]. This is an important finding that links TD to endothelial dysfunction and androgen therapy to endothelial repair. Because endothelial dysfunction contributes to ED, the latter is considered an early warning sign that precedes clinically significant CVD by several years [7,50].
In addition, several clinical studies have supported a role for androgen modulation of endothelial function. In a cross-sectional study, Akishita et al. [23] demonstrated that total and free testosterone levels in men were positively correlated with %FMD, a surrogate marker of clinical atherosclerosis, which also reflects endothelial function [59,60]. Akishita et al. [23] further suggested that total and free testosterone were related to %FMD, independent of age, body mass index, hypertension, hyperlipidemia, diabetes mellitus, and current smoking. Acute [58] and chronic [61] testosterone therapy in men with TD enhanced %FMD without affecting the basal diameter of the brachial artery, which suggests an endothelium-dependent vasodilator action of testosterone. The relationship between %FMD and testosterone was not altered after adjustment for percentage nitroglycerine-mediated dilation (%NMD), which further supports the action of testosterone on endothelial function. In another study of 722 men aged 25 to 85 years, FMD and NMD measurements were carried out and correlated with total and free testosterone levels. It was shown that lower serum total and free testosterone levels are associated with impaired endothelial function [62]. These findings corroborate those reported by Akishita et al. [23] and support the notion that testosterone has a positive role on vascular health [63] via the modulation of endothelial function and endothelial cell repair after injury.
ROLE OF ANDROGENS IN ENDOTHELIUM PROGENITOR CELL DEVELOPMENT, DIFFERENTIATION, MOBILIZATION, AND REPAIR MECHANISMS
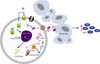
It has been suggested that androgens modulate endothelial cell function and differentiation and the proliferation of EPCs by genomic and nongenomic mechanisms (Fig. 2) [7,45-47]. Testosterone or its active metabolite 5α-DHT bind to the classic AR and elicit genomic responses resulting in increased VEGF and cyclin expression as well as increased eNOS expression and activity. Androgens may also interact with membrane ARs and activate a signaling cascade that results in increased VEGF and matrix metallopeptidase-9 (MMP-9) expression, together with increased activity of eNOS resulting in increased differentiation and proliferation of EPCs from bone marrow stroma precursor cells [64,65].
Foresta et al. [44] investigated the relationship between serum testosterone in patients with hypogonadotropic hypogonadism and circulating levels of both progenitor cells and EPCs. Hypogonadotropic hypogonadism patients exhibited low levels of testosterone and also reduced circulating levels of progenitor cells and EPCs. These observations suggested that testosterone may modulate the differentiation, proliferation, and maturation of these cells. A significant increase in the number of progenitor cells and EPCs was noted subsequent to testosterone treatment [44]. In a subsequent study, the authors demonstrated that the increase in the proliferation, migration, and colony formation activity of EPCs induced by androgens is an AR-dependent pathway [66].
In Klinefelter syndrome patients, the number of circulating EPCs is markedly reduced [67]. Because EPCs have never been studied in this syndrome, the authors evaluated the number of circulating EPCs in 68 adult men with this syndrome (47, XXY) and in 46 healthy men. Patients and controls were divided into two subgroups according to the absence or presence of cardiovascular risk factors [67]. Controls without cardiovascular risk factors had significantly higher levels of EPCs than did controls with cardiovascular risk factors. By contrast, Klinefelter syndrome patients without cardiovascular risk factors had EPC levels similar to those of men with Klinefelter syndrome with risk factors and significantly lower than those of controls without cardiovascular risk factors [67]. The number of EPCs in patients with TD was not different from the number in patients with normal testosterone levels. Twenty-two patients with TD were reevaluated after 6 months of testosterone therapy, but the authors did not observe any modification in the number of EPCs [67]. These findings are contrary to those reported previously in hypogonadal men [44], which suggests that TD associated with Klinefelter syndrome may be confounded by other factors owing to the genetic nature of this disorder.
It should be noted that studies of low serum testosterone and its relation to EPCs vary in quality, with inconsistent findings. Because EPCs express the AR, it seems reasonable to suggest that androgens may influence EPC proliferation and migration. Some studies have reported that men with TD exhibited reduced total basal testosterone, estradiol (E2), luteinizing hormone (LH), and follicular stimulating hormone (FSH) levels, and demonstrated increased numbers of circulating EPCs in response to testosterone therapy [44]. After treatment, no changes in FSH or LH levels were noted; however, both E2 and testosterone levels had increased to normal, physiological values. To ascertain the role of testosterone in modulating EPCs, immunocytochemistry on cultured EPCs showed a stronger signal for AR expression in the nucleus and cytoplasm, which suggests a possible role of androgens in EPC proliferation. The magnitude of the response found may be linked to the restoration of normal testosterone levels given the demonstrated wide expression of AR in EPCs or it may be a corroboration of testosterone conversion to E2 by the enzyme aromatase. However, because both testosterone and E2 levels increased after testosterone administration, the peripheral conversion of testosterone to E2 could not be excluded as a reason for the increased EPC levels. As a follow-up study, EPCs were treated with a synthetic, nonaromatizable androgen in vitro and demonstrated increased migration and proliferation by an AR-mediated mechanism [66]. These findings provide strong support for a role of androgens in stimulating EPC generation, differentiation, and migration from the bone marrow stromal cells. Studies by Fadini et al. [65] demonstrated that androgens exerted no direct effect on late EPCs in vitro or in vivo and noted that androgen stimulation was only observed in early EPCs; no effects on late EPCs were noted.
DISCUSSION
Endothelial dysfunction is a major contributing factor to CVD and represents a shift from a healthy endothelium to a damaged procoagulative, proinflammatory, and provasoconstrictive endothelium [35]. An intact functional endothelium is critical in maintaining the vascular functions of arterial relaxation and venous constriction [68,69]. Endothelial dysfunction contributes to vascular stiffness, increased vascular tone, production of inflammatory cytokines, increased permeability, decreased endothelial cell growth, and dysregulation of fibrinolytic factors [22]. In addition, endothelial dysfunction results in reduced expression and activity of eNOS and increased production of ADMA, a competitive inhibitor of eNOS. Interestingly, in patients with idiopathic hypotrophic hypogonadism, elevated plasma ADMA levels are associated with a reduction in NO production and parenteral testosterone administration reduces ADMA concentrations and increases NO production, which suggests that androgens restore endothelial function [70].
Furthermore, endothelial dysfunction is also associated with increased production of ROS; increased synthesis and release of ET-1, E-2, and E-3; increased production of inflammatory cytokines such as TNF-α; increased expression of markers of cell adhesion such as E-selectin, ICAM, and VCAM; deregulation of fibrinolytic factors such as vWF; inability to regenerate endothelium from EPCs; increased endothelial cell apoptosis; increased cellular/vascular permeability; and increased vascular tone, as noted previously [22]. These pathophysiological mechanisms, coupled with a reduction in EPCs, could underpin a common pathogenesis for CVD and ED and endothelial dysfunction [22,21].
The balance between endothelial repair mechanisms and the extent of injury due to vascular insults may determine the degree or severity of vascular dysfunction. It has been postulated that circulating EPCs are integral to the repair process of the injured vascular beds. The degree of oxidative stress, inhibition of eNOS activity, and increased levels of ADMA and hyperhomocysteinemia are among some of the factors that contribute to endothelial damage. These same factors may impair vascular repair by altering the development, differentiation, or mobilization of EPCs. Therapeutic approaches that modify the vascular risk factors, reduce endothelial injury, and facilitate endothelial repair pathways, such as statin therapy [33,71,72] or androgen replacement therapy, in men with TD have shown a marked increase in circulating EPCs [15,44,65]. These studies suggest that EPCs play a pivotal role in vascular homeostasis and that androgens may have a profound role in modulating EPC differentiation, proliferation, and maturation. Furthermore, these studies suggest that androgens play a role in maintaining endothelial cell function and repair mechanisms.
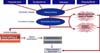
Aging and associated comorbidities contribute to endothelial dysfunction by reducing the ability to protect the endothelium from injury and insult produced by dyslipidemia, insulin resistance, type 2 diabetes, inflammation, metabolic syndrome, obesity, hypertension, and oxidative stress [73]. Because many of these comorbidities are also associated with reduced testosterone levels [73,74], it is possible that TD is a major risk factor for endothelial dysfunction in men [49]. Fig. 3 relates TD and several comorbidities as they relate to endothelial injury. As men age, serum testosterone concentrations decrease by about 1% per year [23]. However, in the current literature, it is not fully understood whether this decrease in testosterone levels is mainly due to normal aging or whether it is related to hypogonadism, a decrease in the functionality of the hypothalamic-pituitary-gonadal axis. Recent studies have suggested that aging per se is not the risk factor for TD and that other risk factors such as obesity and metabolic syndrome may be the real contributors to hypogonadism [75,76]. Testosterone has important metabolic actions in men, including body composition, insulin sensitivity, and lipid metabolism [49,52,69,77,78]. Low testosterone levels are also associated with ED [48,49,77]. This is important because ED is a vascular condition, and because ED and CVD are closely related, ED can be the first clinical manifestation of underlying CVD and can serve as an early warning sign for primary care physicians [23,49,79]. Thus, it is possible to infer that reduced levels of testosterone negatively impact vascular dynamics and the reactivity of blood vessels (Fig. 4) [9,49,77,78].
The actions of regenerating and repairing areas of vascular insult due to ischemia or endothelial injury do not depend solely on the cells residing in the area of insult but are influenced by bone marrow cells [27,33,72,80]. EPCs are premature, circulating, bone marrow-derived cells with putative trans-differentiation potential, as was shown in Figs. 1, 2 [80]. The risk factors thought to be involved are illustrated in Fig. 4 and may lead to a decreased number of circulating EPCs [33,80]. Studies have also shown that men with CVD risk factors are likely to have lower testosterone, which is independently associated with endothelial dysfunction [7]. Men with congestive heart disease exhibit a high prevalence of low testosterone levels, irrespective of age [49]. Araujo et al. [81] suggested that cardiovascular mortality via endothelial dysfunction is driven by the underlying health status and that a low testosterone level is a marker of poor general health; however, low testosterone and disease pathology exist as concomitant risk factors [81]. Inflammation, obesity, and insulin resistance are known to reduce testosterone levels, and testosterone is known to confer beneficial effects on those risk factors, thus ameliorating CVD [20,23,49,52,77,78].
Reduced levels of testosterone may exacerbate obesity, insulin resistance, dyslipidemia, and hypertension, among others [7,49,77-79]. In recent studies, testosterone treatment for ED ameliorated metabolic syndrome components (such as insulin resistance) and improved endothelial function [48]. Furthermore, the administration of testosterone undecanoate therapy to hypogonadal men with ED and venous leakage ameliorated erectile function by improving veno-occlusion [48,82]. Testosterone may have a beneficial effect on the systemic vasculature by ameliorating ED, thus reinforcing the idea that testosterone supplementation could prevent or delay the progression of disease [7].
The Massachusetts Male Aging Study [23,83] confirmed that ED is highly correlated with coronary artery disease, hypertension, heart disease, and diabetes. Men with low levels of testosterone and without CVD also have decreased blood flow and reduced nitrate-dependent vasodilation [84]. Zitzmann [51] found that the number of the trinucleotides comprised of the repeat of cytosine, adenine and guanine repeats in the AR, which reduces the sensitivity to testosterone, was associated with endothelium-dependent and endothelium-independent brachial artery vasodilation in men, which shows the effect of the AR action on vasoreactivity [49].
The common denominator among the underlying pathophysiology of ED and CVD is vascular insufficiency promoted by atherosclerosis [85,86] via endothelial dysfunction and oxidative stress from various metabolic conditions [24]. An increase in circulating EPCs is thought to be related to the bioavailability of NO, including increased activity of eNOS, anti-inflammatory and antioxidative enzymes, and the activation of MMP-9 [87]. Furthermore, because testosterone deprivation decreases the availability of NO [66], and because NO is a key marker of endothelial function, it can be inferred that testosterone has a direct effect on the endothelium or circulating EPCs via an increase in NO [66,88]. Burger and Touyz [35] showed that healthy endothelial cells possess certain cellular biomarkers, and dysfunctional endothelium exhibits impaired vasodilator synthesis and release, such as of NO and PGI2, and an increase in ROS [35]. Yu et al. [89] have also shown the benefits of testosterone in activated eNOS in vascular endothelial cells, thereby increasing the amount of NO in the endothelium.
Additionally, Hamed et al. [25] showed that men with type 2 diabetes showed a positive correlation between circulating NO and circulating EPCs. This is in direct affirmation of Burger and Touyz [35]'s study, which stated that a dysfunctional endothelium has low concentrations of NO. Hyperglycemia produces ROS, which then decrease NO availability [25]. Hyperglycemia can conjure ROS buildup via increased PKC/NADPH oxidase activity, which can lead to an uncoupling of eNOS and reduced NO availability, thus impairing the number of progenitor cells and their subsequent function [25]. It is known that deregulation of vascular tone, or endothelial dysfunction, is characterized by decreased availability of NO, which inadvertently induces the immune system to release white blood cells to the site of vascular deregulation [90]. This is of particular importance because of the role testosterone plays in NO production and maintenance. Shabsigh et al. [53] demonstrated in animal studies that the expression of various NOS isoforms in the corpora cavernosum is regulated by androgens. Also, NOS activity and protein expression are decreased in castrated animals, and testosterone administration restores and normalizes NOS protein expression and activity, allowing mobilization and homing of EPCs to areas of vasculogenesis and angiogenesis [53].
There remains an inextricable link between the role of testosterone and ED; however, this link provokes some controversy owing to the multifactorial nature of the pathophysiology of ED. Testosterone is critical to the health of the vascular beds [49,56], stimulating endothelial cell proliferation via an AR/VEGF-mediated mechanism, and promoting angiogenesis [56]. Furthermore, because testosterone deprivation is known to decrease the availability of NO [66], and because NO is a key marker of endothelial function, it can be inferred that testosterone may have a direct effect on endothelial function and development and the differentiation, maturation, migration, and homing of circulating EPCs.
Although the role of androgens in modulating endothelial function and in the development and differentiation of progenitor cells is not fully understood, several studies have shown that testosterone has a positive effect on the endothelium and improves the number of EPCs. More molecular and biochemical studies are warranted to delineate the exact mechanisms by which testosterone regulates endothelial function. The arena of EPCs and their role in vascular repair and treatment of disease is an emerging field, and the role of testosterone in this physiological process will require more detailed investigations. Cellular and molecular understanding of these processes will unlock new therapeutic approaches for the treatment of ED and CVD in men.

 XML Download
XML Download