

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Wilms' tumor (WT) is the most common primary malignant renal tumor in childhood. The reported prevalence of WT is 8 cases for every 1 million children [1]. Numerous anomalies and syndromes have been associated with WT. Many of these involve the genitourinary tract, including cryptorchidism, male pseudohermaphroditism, hypospadias, and renal anomalies such as hypoplasia, ectopia, duplication anomalies, and horseshoe kidney (HK) [2]. There is a tendency to develop neoplasia in HKs, with hypernephroma being seen most commonly, followed by renal pelvis tumors and WT [3].
From 1969 to 1998, 8,617 patients were enrolled in the National Wilms Tumor Study Group (NWTSG). Forty-one patients were found to have a WT arising in a HK, an incidence of 0.48%, but HK was not recognized preoperatively in 13 patients [4]. Sometimes it is not feasible to detect WT in a HK before treatment.
We present a case of WT associated with a HK in a child that was successfully managed with preoperative chemotherapy followed by surgical resection and adjuvant chemotherapy to preserve renal function.
CASE REPORT
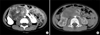
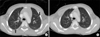
A healthy 5-year-old boy presented with an asymptomatic abdominal mass that had been noted by his mother. On abdominal physical examination, two fist-sized masses were palpated in the right flank and umbilical region. On palpation, the mass was relatively nonmobile, nontender, and hard in consistency. The results of laboratory blood and urinary analyses were within normal limits and the urinary excretion of vanillylmandelic acid, catecholamines, and homovanillic acid was also within the normal range. An abdominal computed tomography (CT) scan confirmed an 11 cm×7 cm mass that appeared to arise from the isthmus of a HK. A band of renal parenchyma was seen extending across the midline, which was suggestive of a HK (Fig. 1A). A post-CT abdominal X-ray film revealed bilateral malrotation typical in a HK and bilateral hydronephrosis caused by extrinsic ureteral compression of the mass. Even though the results of a chest X-ray film were normal, a chest CT scan showed two small nodules in the left upper lobe anterior segment and right upper lobe posterior segment of the lung (Fig. 2A). Fine-needle biopsy of the mass revealed the presence of blastema, stromal, and epithelial cells without anaplasia, which suggested a diagnosis of WT with favorable histology (Fig. 3).
The patient was classified as having a stage IV WT on the basis of the presence of metastatic small nodules in the chest and was treated with 6 weeks of neoadjuvant chemotherapy. We used the DD-4A regimen (pulse-intensive dactinomycin, vincristine, and doxorubicin) under the proposed Children's Oncology Group (COG) protocol (AREN0533). An abdominal CT scan revealed a decrease in tumor size from 11×7 cm to 8×5 cm and central necrotic changes (Fig. 1B), and a chest CT scan after 6 weeks of neoadjuvant chemotherapy showed disappearance of the small nodules in the lungs (Fig. 2B). The patient's abdomen was thereafter explored by a transperitoneal approach. There was a hard, spherical mass involving the isthmus of the HK with no infiltration of the surrounding structures. There was no paraaortic lymphadenopathy and the inferior vena cava, liver, and spleen were normal. Resection of the tumor was done with an isthmusectomy and bilateral partial nephrectomy of the lower poles. The pathologic report of the resected tumor showed 30% necrosis of the whole tumor tissue and the resection margins were free of tumor (Fig. 3). The perioperative serum creatinine did not change and was 0.6 mg/dl. The patient completed 25 weeks of adjuvant chemotherapy according to the DD-4A regimen postoperatively.
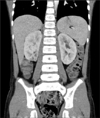
At the end of the treatment, there were no signs of any recurrent disease on the CT scans or positron emission tomography scans and the bilateral hydronephrosis had disappeared (Fig. 4).
DISCUSSION
Assuming the incidence of HK in the general population is 1 in 400, a child with a HK has a 2-fold increased risk of having WT compared with the general population [4]. In Korea it is also rare; only one case of WT in a HK has been reported in the Korean Journal of Urology [5]. A high index of suspicion should be maintained, and routine surveillance should be performed on HK patients. Although the etiology of WT in HK is unknown, some speculate that WT develops as a result of sequestered metanephric blastemas in the isthmus, which harbor malignant potential. Others have hypothesized that the embryologic lesion that results in a HK may predispose the kidney to a second event resulting in WT [4].
Children with WT in a HK harbor problems in diagnosis, and it is not uncommon for the diagnosis to be established at the time of surgery. Of the 41 cases of WT in a HK, the HK was not recognized preoperatively in 13 patients (32%) [4]. In our case, a band of renal parenchyma extending across the midline that was seen on the abdominal CT scan, the malrotation of the kidneys, and the bilateral hydronephrosis on post-CT abdominal X-ray films were helpful in diagnosing the HK. CT is a reliable method for diagnosing WT in a HK. Accurate preoperative diagnosis of a HK in WT is important for planning treatment modalities and may help to decrease complications related to transection of the urinary collecting system.
Multimodality treatment has been used to treat WT successfully. Multiple clinical trials have been conducted by the NWTSG (now incorporated into the COG) and the International Society of Pediatric Oncology (SIOP) to determine the appropriate role of the therapeutic modalities available. Stage and histopathology are the most important determinants of outcome in children with WT. There are currently two staging systems available reflecting treatment differences [6]. The system used by the COG reflects staging at primary surgery. Alternatively, the staging by the SIOP is performed after preoperative chemotherapy. One of the main controversies in the treatment of children with WT is whether to introduce preoperative chemotherapy, as suggested by the SIOP. Opponents of preoperative chemotherapy have argued that the preoperative treatment leads to either overtreatment or undertreatment owing to incorrect staging and histological evaluation. Proponents of preoperative therapy suggest that the tumor is easier to resect with a decreased incidence of tumor spillage and lower mortality and morbidity [6]. Despite the arguments, specific patients seem to benefit from preoperative chemotherapy; these include patients with bilateral WTs, those with inferior vena cava involvement, and patients with massive tumors that are unresectable without undue risk to the patient [6]. The use of neoadjuvant chemotherapy can mean that patients who do not have a confirmed diagnosis of WT may receive unnecessary therapy. The error rate in SIOP-9 was 5% (28/511), and of this group, 20 patients had another malignant tumor type, whereas 8 had benign pathology [7]. The resolution to this particular argument could be provided by a pre-chemotherapy biopsy. In our case, we chose percutaneous fine-needle biopsy and preoperative chemotherapy owing to the large tumor size in the HK to decrease surgical complications. In the COG protocol (AREN0533), patients with stage IV disease without loss of heterozygosity in 1p and 16q whose pulmonary lesions respond "rapidly and completely" are treated with the DD-4A chemotherapy regimen and no pulmonary irradiation. In this patient, chromosomal study showed no abnormal findings and the small metastatic lung nodules completely disappeared after preoperative chemotherapy. Thus, the patient completed the DD-4A chemotherapy regimen after complete resection of the tumor without lung irradiation.
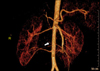
There is great variation in the shape of a HK, with a variable relationship between the great vessels and the ureter. In 30% of cases, there is one renal artery to each kidney. Duplicated or even triplicated renal arteries may supply one or both kidneys. The blood supply to the isthmus may come from the renal artery, directly from the aorta, from the inferior mesenteric artery, or from the iliac arteries [8]. Angiography might be helpful in diagnosing a tumor as well as in planning the excision and mapping the blood supply in WT arising in a HK. In this case, we reconstructed CT angiography before surgical excision. The CT angiography revealed two renal arteries in both kidneys and the blood supply of the tumor came directly from the aorta through the isthmus (Fig. 5).
Most authors recommend removal of the involved kidney and the isthmus for WT in a HK [9]. Resection of the isthmus is important because if the urine does not drain through the remaining kidney, a urinary fistula may result. If the tumor arises in the isthmus itself, bilateral lower pole heminephrectomies and isthmusectomy should be performed [10].
Of the 41 cases of WT in a HK, 37% of patients were judged to be inoperable cases at the time of initial exploration but all were amenable to resection after chemotherapy [4]. Resection of WT in a child with a HK presents unique challenges. The use of preoperative chemotherapy in this unusual combined clinical presentation may offer the advantage of more conservative surgery, may decrease surgical morbidity resulting from tumor spillage and incomplete surgical resection, and may preserve more renal function after surgical excision in a child. The overall survival of patients with WT in a HK appears to be about the same as for WT in normal kidneys. The estimated survival 4 years after diagnosis of 41 patients was 86% in the NWTSG report [4].
In conclusion, children with HK and WT must be carefully examined before any surgery by use of CT. Preoperative chemotherapy in this condition might be a good treatment method for decreasing surgical morbidity, promoting complete excision, and preserving renal function.

 XML Download
XML Download