

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Peyronie's disease (PD) is a relatively common condition and affects 3.2 to 8.9% of the male population [1,2]. PD is characterized by severe fibrotic processes in the tunica albuginea, which hinders expansion of the tunica albuginea and results in penile curvature [3]. The etiology of PD is not yet fully uncovered. Recent studies suggest that localized inflammatory processes and aberrant wound healing in the tunica albuginea following minor trauma to the penis during sexual intercourse are responsible for the plaque development [4,5]. However, the available medical treatment options, including oral, intralesional injection, and topical therapies, have failed to demonstrate conclusive effects, and surgical treatment is currently the only efficacious treatment for PD [6-8]. It is necessary to understand the pathophysiologic mechanisms of PD at the cellular and molecular levels to develop targeted and curative treatment modalities for PD.
Overexpression of profibrotic cytokines is known to play an important role in the development of fibrotic plaque, and transforming growth factor-β1 (TGF-β1) is the most extensively studied as a candidate fibrogenic cytokine. We and other investigators have reported that the expression and activity of the TGF-β1 and Smad transcription factors are significantly increased in human PD plaque and in fibroblasts derived from patients with PD [9-11]. Moreover, we observed that local injection of adenovirus encoding the TGF-β1 gene (ad-TGF-β1) into the tunica albuginea of rats results in histologic and morphological changes in the tunica that are similar to those found in human PD plaques [12], which further supports a causative role of TGF-β1 in the pathogenesis of PD. Therefore, targeted inhibition of the TGF-β signaling pathway might be a valuable therapeutic strategy for the treatment of PD. Recently, small-molecule inhibitor of TGF-β type I receptor, activin receptor-like kinase 5 (ALK5) inhibitor, was shown to decrease fibrosis in kidney, lung, and liver [13-15]. We also recently found that local injection of an ALK5 inhibitor, 3-((5-(6-Methylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)methyl)benzamide (IN-1130), into the tunica albuginea induced the regression of tunical fibrosis and corrected penile curvature in a rat model of PD induced by repeated intratunical injection of ad-TGF-β1 [16].
In the present study, we determined the effectiveness of IN-1130 on the TGF-β1-induced activation of the Smad transcription factors and extracellular matrix production in fibroblasts isolated from human PD plaque.
MATERIALS AND METHODS
1. Cell culture and characterization
Plaque tissue from a patient with PD was used for primary fibroblast culture. Plaque tissue was transferred into sterile vials containing Hank's balanced salt solution (Invitrogen, Carlsbad, CA, USA). After washing three times in phosphate-buffered saline (PBS), the tissue was minced into 1 mm2 segments and incubated in Dulbecco's modified Eagle Medium (DMEM) supplemented with 0.06% collagenase A (Sigma-Aldrich Co., St. Louis, MO, USA) for 1 hour. After centrifugation (400 g × 5 minutes) and washing in fresh culture medium, the cells and tissue fragments were collected and placed in 100 mm cell culture dishes (BD, Franklin Lakes, NJ, USA) under standard conditions using DMEM supplemented with 10% fetal calf serum, penicillin (100 U/ml), and streptomycin (100 µg/ml). The dishes were then incubated in a humidified 37℃ incubator with 5% CO2. The study protocol was approved by the Institutional Review Board of our university.
For the characterization of primary cultured cells, the cells were cultured on sterile cover glass until nearly confluent. After serial washes with PBS, the cells were fixed in 4% paraformaldehyde for 10 minutes at 4℃ and in 100% methanol for 10 minutes at 4℃. Individual chambers were incubated with antibody to vimentin (1:500; a fibroblast marker, Sigma-Aldrich Co.), smooth muscle α-actin (1:500; a myofibroblast marker, Sigma-Aldrich Co.), desmin (1:500; a smooth muscle cell marker, Abcam, Cambridge, UK), or platelet/endothelial cell adhesion molecule (PECAM-1, an endothelial cell marker; Chemicon, Temecula, CA, USA; 1:500) for 1 hour at room temperature. After serial washes with PBS, the chambers were incubated with fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse immunoglobulin (IgG) (1:1,000; Zymed Laboratories, South San Francisco, CA, USA) or FITC-conjugated goat anti-hamster IgG (1:1,000; Jackson ImmunoReseach Laboratories Inc., West Grove, PA, USA) for 2 hours at room temperature. For the nuclei labeling, medium containing 4,6-diamidino-2- phenylindole (DAPI, Vector Laboratories Inc., Burlingame, CA, USA) was applied to the chamber.
2. Western blot
IN-1130, an ALK5 inhibitor, was provided by SK Chemicals (Seongnam, Korea). After serum starvation for 24 hours, the fibroblasts were pretreated for 1 hour with 10 µM IN-1130 (molecular weight, 518). For detection of the P-Smad2, total Smad2, P-Smad3, and total Smad3 proteins, the fibroblasts were treated with 10 ng/ml TGF-β1 (R&D Systems Inc., Minneapolis, MN, USA) for 1 hour, and for detection of the plasminogen activator inhibitor-1 (PAI-1), fibronectin, collagen I, and collagen IV proteins, the cells were incubated with TGF-β1 for 24 hours.
Cytosolic and nuclear proteins were extracted by using a commercially available kit (NE-PER Nuclear and Cytoplasmic Extraction kit; Pierce Biotechnology, Rockford, IL, USA). Briefly, after serial washes with PBS, the cells were harvested and centrifuged for 5 minutes at 3,000 rpm (4℃). Cell pellets were incubated in 100 µl cytoplasmic extraction reagent I on ice for 10 minutes, and then 5.5 µl cytoplasmic extraction reagent II was added to the sample and incubated on ice for 1 minute. After centrifugation for 5 minutes at 13,000 rpm (4℃), the supernatant (cytosolic extract) was transferred to a tube on ice. Pelleted nuclei were resuspended in 100 µl nuclear extraction reagent on ice. After serial vortexes for 15 seconds every 10 minutes on ice for 40 minutes, the samples were centrifuged for 10 minutes at 13,000 rpm (4℃) and the supernatant (nuclear extract) was transferred to a tube on ice. Equal amounts of each cytosolic and nuclear protein fraction (80 µg/lane) were separated by 10% sodium dodecylsulfate-polyacrylamide gel electrophoresis and immunoblotting. After protein transfer, nitrocellulose membranes were incubated with antibody against P-Smad2, total Smad2 (which also recognizes Smad3), P-Smad3 (1:300; Cell Signaling, Technology Inc., Danvers, MA, respectively), total Smad3 (1:300; Zymed Laboratories Inc.), Hsp90 (1:300; a cytosol marker, Abcam plc), or lamin (1:50; a nuclear marker, Abcam plc).
Equal amounts of protein from whole-cell extracts (80 µg/lane) were separated by 10% sodium dodecylsulfate-polyacrylamide gel electrophoresis and immunoblotting. After protein transfer, nitrocellulose membranes were incubated with antibody against PAI-1 (1:600; Abcam plc), fibronectin (1:300; Abcam plc), collagen I (1:300; Abcam plc), collagen IV (1:300; Abcam plc), or β-actin (1:6,000; Abcam plc). Results were quantified by densitometry.
3. Nuclear translocation
The fibroblasts were cultured on sterile cover glass until nearly confluent. After serum starvation for 24 hours, the cells were pretreated for 1 hour with 10 µM IN-1130 and were then treated with 10 ng/ml TGF-β1 (R&D Systems Inc.) for 1 hour. Individual chambers were processed as described above and were then incubated with antibody to total Smad2 (which also recognizes Smad3, 1:500; Cell Signaling Inc.) for 3 hours at room temperature. After several washes with PBS, the chambers were incubated with FITC-conjugated goat anti-mouse IgG (1:1,000; Zymed Laboratories Inc.) for 2 hours at room temperature. For the nuclei labeling, medium containing DAPI (Vector Laboratories Inc.) was applied to the chamber.
Digital images were captured with an Apotome microscope (Zeiss, Göttingen, Germany), and fluorescent intensity was measured for every nucleus in the field and averaged for the field with an image analyzer system (National Institutes of Health Image J, Bethesda, MD, USA).
RESULTS
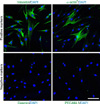
1. Isolation and characterization of primary cultured fibroblasts derived from human PD plaque
More than 95% of primary cultured cells showed positive staining for fibroblast marker (vimentin) but did not reveal positive staining for antibody to desmin (smooth muscle cell marker) or PECAM-1 (endothelial cell marker) (Fig. 1). Similar to the result from a previous study [17], about 20% of the primary cultured cells stained positive for antibody to smooth muscle α-actin (myofibroblast marker) (Fig. 1).
2. Effect of IN-1130 on TGF-β1-induced phosphorylation of Smad2 and Smad3 in fibroblasts derived from human PD plaque
We determined the ability of IN-1130 to inhibit TGF-β1-induced Smad2 and Smad3 phosphorylation. At 1 hour after treatment with recombinant TGF-β1 protein, we observed a profound increase in phosphorylation of Smad2 and Smad3 in both cytosolic and nuclear fractions. Preincubation with IN-1130 substantially inhibited phosphorylation of Smad2 and Smad3 (Fig. 2).
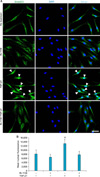
3. Effect of IN-1130 on TGF-β1-induced nuclear translocation of Smad2/3 in fibroblasts derived from human PD plaque
TGF-β1 is known to be involved in the nuclear shuttling of Smad2 and Smad3, i.e., the translocation of Smad proteins from the cytoplasm to the nucleus [18]. Therefore, we asked whether ALK5 activity is necessary for TGF-β1-induced nuclear translocation of Smad2/3. Fluorescent immunocytochemistry of fibroblasts with antibody against total Smad2 (which also recognizes Smad3) and nuclei labeling with DAPI revealed that treatment with IN-1130 significantly reduced TGF-β1-induced nuclear accumulation of Smad proteins (Fig. 3).
4. Effect of IN-1130 on TGF-β1-induced extracellular matrix production in fibroblasts derived from human PD plaque
To evaluate the effect of ALK5 inhibition on extracellular matrix production, we examined the effect of IN-1130 on TGF-β1-induced PAI-1, fibronectin, collagen I, and collagen IV expression in fibroblasts. Treatment of fibroblasts with TGF-β1 significantly increased the production of extracellular matrix proteins. IN-1130 significantly inhibited TGF-β1-induced production of extracellular matrix markers. IN-1130 did not affect the basal production of extracellular matrix proteins in fibroblasts not stimulated with TGF-β1 (Fig. 4).
DISCUSSION
In this study, we showed that a small-molecule inhibitor of ALK5, IN-1130, successfully blocked TGF-β1-induced signaling, i.e., phosphorylation and nuclear translocation of Smad2 and Smad3, and inhibited extracellular matrix production in fibroblasts derived from human PD plaque.
TGF-β-mediated fibrotic responses begin by activating the receptor-associated Smads, including Smad2 and Smad3 [19]. ALK5 is a TGF-β type I receptor specifically involved in the activation of TGF-β. TGF-β induces the phosphorylation of serine/threonine residues of ALK5 and then phosphorylates the major downstream signaling molecules Smad2 and Smad3. Phosphorylated Smad2 and Smad3 form a heteromeric complex with Smad4 and translocate into the nucleus. When translocated, phospho-Smad2 and phospho-Smad3 regulate the transcription of TGF-β-responsive genes and induce tissue fibrosis [19]. Previous studies revealed an activation of Smad2 or Smad3 in the kidneys of diabetic animals [20,21], and targeted deletion of the Smad3 gene attenuated diabetes-induced renal fibrosis [22], which supports a major role of Smad2 and Smad3 in renal fibrosis. The activation of Smad2 and Smad3 in human PD plaque or in fibroblasts isolated from a PD patient as shown by us [11] and other investigators [10] indicates that inhibition of the TGF-β pathway may be a promising therapeutic strategy for PD.
The phosphorylation of Smad2 or Smad3 is necessary for nuclear translocation and TGF-β-mediated fibrosis [23]. In the present study, IN-1130 substantially inhibited TGF-β1-induced Smad2 and Smad3 phosphorylation and the nuclear shuttling of Smad proteins. The ALK5 kinase activity affects nuclear translocation and accumulation of Smad transcriptional factors [24-26], which is responsible for activating the profibrotic genes that are involved in the production and deposition of extracellular matrix protein. Therefore, we examined whether IN-1130 can prevent the stimulatory effect of TGF-β1 on the production of extracellular matrix markers. A previous study of newborn foreskin fibroblasts and dermal fibroblasts showed that an ALK5 inhibitor, SB431542, inhibited the TGF-β1-induced expression of extracellular matrix protein [27]. Similarly, IN-1130 also significantly reduced TGF-β1-induced production of PAI-1, fibronectin, collagen I, and collagen IV, which was comparable to the basal levels.
In the present study, we determined the antifibrotic effect of a selective ALK5 inhibitor, IN-1130, in human PD fibroblasts in vitro. The current results showing the efficacy of IN-1130 in primary cultured fibroblasts derived from human PD and our recent results in a PD animal model in vivo suggest that ALK5 inhibition may represent a promising option for treating this condition [16].
CONCLUSIONS
A small-molecule inhibitor of ALK5, IN-1130, successfully blocked TGF-β1-induced Smad2/3 activation and extracellular matrix production in primary fibroblasts derived from human PD plaque. Overexpression of TGF-β and activation of Smad transcriptional factors play a crucial role in the pathogenesis of PD. Thus, inhibition of TGF-β signaling pathway will be a promising therapeutic strategy for treating PD.


 XML Download
XML Download