

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The newborn infant with ambiguous external genitalia presents a problem of sex assignment and is frequently described as a clinical emergent situation that is distressing to the parents. The word intersex has conventionally been used to refer to the appearance of the external genitalia being at variance with normal development for either sex; however, this term is not favored by many families with ambiguous genitalia [1]. A multidisciplinary meeting of medical and nonmedical experts in Chicago in 2005 (the Chicago Consensus) established revised nomenclature and treatment recommendations in individuals with the newly defined term disorders of sex differentiation (DSDs) replacing terms such as intersex, hermaphroditism, and pseudohermaphroditism [2-4]. DSDs are defined as congenital conditions associated with atypical development of chromosomal, gonadal, or anatomical sex [4]. There are limited data on the incidence of DSDs; it is estimated that the overall incidence of DSDs is one in 5,500 [5,6]. Congenital adrenal hyperplasia (CAH) and mixed gonadal dysgenesis are the most common causes of ambiguous genitalia, constituting approximately over 50% of all cases of genital ambiguity in the newborn period [7]. The incidence of CAH and mixed gonadal dysgenesis worldwide is 1:15,000 and 1:10,000, respectively, but varies considerably among different populations [8,9]. Confirming a cause for DSD and devising a management plan is one of the most challenging clinical conditions for the pediatric urologist. It is important to diagnose DSDs correctly as soon as possible to counsel the parents appropriately. The evaluation and management of DSDs is complex, and a multidisciplinary team approach including a pediatric urologist, a psychiatrist, and a pediatric endocrinologist is required for optimal management [4,10-12], with communication with the primary care physician [13]. This article is an attempt to outline pragmatic perspectives in the approach to patients with DSDs.
NOMENCLATURE AND CLASSIFICATION OF DSD
1. Dilemma of the preexisting nomenclature
Advances in understanding the molecular genetic causes of abnormal sexual development and heightened awareness of the ethical and patient-advocacy issues mandated a reexamination of preexisting nomenclature [11]. Terminologies such as intersex, hermaphroditism, and pseudohermaphroditism are controversial; potentially pejorative to patients; and confusing even to urologists [1]. Therefore, the term DSD was proposed to indicate congenital conditions with atypical development of chromosomal, gonadal, or anatomic sex. A classification is proposed in which DSDs associated with sex chromosome abnormalities (sex chromosome DSD) were separated from DSDs with a normal chromosome complement (46,XX DSD and 46,XY DSD). The stigmatizing term intersex has thus been replaced by a more general and descriptive term, DSD.
2. New classification based on Chicago consensus
Table 1 summarizes the new taxonomy and Table 2 shows the application of the new nomenclatures in clinical situations. Although DSD may be useful as a more global term, these general subcategories seem too nonspecific and less useful to specific clinical situations. Adding diagnostic specificity to the comprehensive DSD definition utilizes knowledge of the karyotype, which is based on recognizing the central role of karyotype analysis in the investigation of DSDs. Subsequently, the confusing mythological term pseudohermaphroditism is replaced. The original summary publications of the Chicago Consensus have not provided precise classifications of the DSDs; therefore, inconsistency exists between the DSD classifications used by each investigator. Further clinical classification based on a primary genetic defect is preferred when available because these could more clearly predict disease-specific outcomes. Although there are potential criticisms to the new nomenclature, the DSD terminology has been generally accepted and is now popularly used in the literature. This term DSD is seen in conferences, in the scientific literature, and even in text books of endocrinology. For example, the International Society for Hypospadias and Intersex Disorders titled its World Congress "Hypospadias and Disorders of Sex Development."
CLINICAL EVALUATION OF DSD
The birth of a child with ambiguous external genitalia is highly distressing to families. The first question parents ask about their newborn is whether it is a boy or a girl. The birth of a newborn with ambiguous genitalia comes as a surprise for the parents and doctors alike. Although some researchers report that 60% of affected children are diagnosed prenatally, many parents are faced with the situation at birth [14]. Healthcare professionals involved in the care of a child with DSD should emphasize to families that a child with DSD has the potential to become a well-adjusted, functional member of society [15].
1. History
An initial approach begins with a complete family and prenatal history taking [4,12]. A detailed history from the parents is needed that is especially focused on the following: ambiguity, hirsutism, precocious puberty, amenorrhea, infertility, unexplained sudden infant death, or consanguinity. Additionally, maternal exposure to hormones including exogenous hormones used in assisted reproductive techniques and the maternal use of oral contraceptives during pregnancy must be assessed [16-21]. Prenatal androgen exposure is clearly associated with psychosexual development [16,17].
2. Physical examination
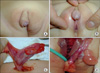
History taking should be combined with a general physical examination with special attention to the genital anatomy [4,12]. Any abnormal virilized or cushingoid appearance of the mother should be checked. It is important to examine the groin and scrotal or labial folds to determine the presence of palpable gonads. For differential diagnosis and treatment purposes, the presence of one or two gonads is an important finding. A palpable gonad is highly suggestive of a testis or rarely an ovotestis (Fig. 1), because the ovaries and streak gonad do not descend. An abnormal phallic size should be noted by width and stretched length measurements. Historically considered, phallus size was considered first in the 1960s when any child with a stretched penile length <2.5 cm was likely to be assigned as female regardless of the underlying diagnosis [22]. Through a rectal exam, we can confirm the presence of a uterus and cervix. Physical examination should be done in a warm room and the patient should be placed supine in the frog leg position. It is important to note the size, location, and texture of both gonads, if palpable. The undescended testis could be located in the inguinalcanal, the superficial inguinal pouch, at the upper scrotum, or rarely in the femoral, perineal, or contralateral scrotal regions. All examinations should be done in the presence of the parents, who should be informed exactly what will be done and why. Medical photography requires sensitivity and consent [23].
3. Criteria of physical findings for DSD evaluation
Criteria of physical findings suggestive of DSD include: 1) overt genital ambiguity (e.g., cloacal extrophy); 2) apparent female genitalia with an enlarged clitoris and posterior labial fusion (e.g., CAH); 3) apparent male genitalia with bilateral undescended testes, hypospadias, or micropenis; and 4) discordance between genital appearance and a prenatal karyotype [4]. Most DSDs are diagnosed in the neonatal period. Later presentations in older children often include: 1) previously unrecognized genital ambiguity, 2) inguinal hernia in a girl (e.g., complete androgen insensitivity), 3) delayed or incomplete puberty, 4) primary amenorrhea or virilization in a girl, 5) breast development in a boy, and 6) gross or cyclic hematuria in a boy [4]. Given the spectrum of findings and diagnoses, no specific single protocol could be used in the evaluation of DSD patients.
4. Laboratory tests
The first-line testing in newborns includes karyotyping with X- and Y-specific probe detection to determine the chromosomal sex; imaging (abdominopelvic ultrasound and retrograde genitogram); serum measurements of 17-hydroxyprogesterone, testosterone, gonadotropins, anti-müllerian hormone, and serum electrolytes to rule out salt-wasting CAH; and urinalysis [4,12]. Now many countries screen newborns for CAH by use of filter-paper blood spot 17-hydroxyprogesterone measurements [24]. The testosterone level can help to determine whether the DSD is due to a lack of androgen or cortisone synthesis or rather due to a receptor defect. Once the karyotype is determined, serum measurements will assist in narrowing the differential diagnosis. For example, if the 17-hydroxyprogesterone level is elevated, a diagnosis of CAH can be made. Determining the levels of 11-deoxycortisol and deoxycorticosterone will help to make a differential diagnosis between 21-hydroxylase and 11β-hydroxylase deficiencies. If the levels are elevated, a diagnosis of 11β-hydroxylase deficiency could be made, whereas low levels confirm 21-hydroxylase deficiency.
5. Imaging test
Noninvasive and inexpensive ultrasound could be the first radiologic examination performed. Abdominopelvic ultrasoundis used for the evaluation of female internal organs and to rule out possible adrenal anomalies. Although it has limited accuracy in detecting intra-abdominal testes, ultrasound can detect gonads in the inguinal region and can identify müllerian anatomy. Retrograde genitogram is performed for the anatomical outlining of the urogenital sinus and for localizing the position and entry of the urethra and vagina into the sinus.
6. Hormonal stimulation test
In certain circumstances, additional testing such as human chorionic gonadotropin and adrenocorticotropin stimulation tests to assess testicular and adrenal functions, respectively, are needed. Despite considerable progress in the genetic basis of human sexual development, a specific molecular diagnosis is identified in only 20% of cases of DSDs [25]. Markedly virilized 46,XX DSDs have CAH; 50% of undervirilized 46,XY DSDs receive a definitive diagnosis [26,27]. Although some gene analyses could be performed in clinical service laboratories, gene testing is not routinely performed in children with DSDs for reasons of cost, insurance, and quality controls [28].
7. Diagnostic laparotomy or laparoscopy
In rare cases in which a definite diagnosis cannot be determined and in infants with intra-abdominal or nonpalpable testes in whom DSDs are considered, open or laparoscopic exploration with biopsy of the gonads could become necessary [29]. In some cases, the differential diagnosis of DSD depends on the interpretation of the histologic features of the gonads [30,31]. Infants with intra-abdominal or nonpalpable testes in whom the precise diagnosis is unavailable with karyotyping and serum study will require an open or laparoscopic exploration with bilateral deep longitudinal gonadal biopsies for histologic evaluation, which will determine the presence of ovotestes, streak gonads, or dysgenetic testes, thereby confirming the diagnosis.
GENDER ASSIGNMENT
1. Influencing factors
Initial gender uncertainty is distressful news for families. Thereby, gender assignment in a newborn with ambiguous genitalia is regarded as a medical emergency, and surgical intervention to match the gender as soon as possible after-medical stabilization is recommended. Influencing factors to consider when discussing gender assignment include diagnosis, genital appearance, fertility potential, therapeutic/surgical options, and familial views or circumstances relating to cultural biases [4,32].
2. Gender assignment according to specific diagnosis
When a specific diagnosis can be made, recommendations for gender assignment can be based upon outcome data. The Chicago Consensus statement did not include specific gender assignment recommendations for all diagnoses, but some consensus participants have provided more specific recommendations. For 46,XX CAH, 90% of individuals raised as females maintain an assigned female gender identity and do not feel gender dysphoric [33]. Therefore, there is agreement among healthcare professionals to raise these patients as females [12,34]. All patients with 46,XY complete androgen insensitivity syndrome (CAIS), who are assigned female sex in infancy identify as females and there is agreement to raise these patients as females [32,35]. More than 60% of 5α-reductase-deficient patients assigned female gender in infancy and virilizing at puberty and all assigned males live as males and also half of 17β-hydroxysteroid dehydrogenase-3 deficiency assigned females in infancy ultimately change their gender role [36]. Therefore, it is recommended to discuss with the families when deciding on gender assignment for patients with 5α-reductase deficiency and 17β-hydroxysteroid dehydrogenase-3 deficiency diagnosed in infancy. For patients with partial androgen insensitivity syndrome, androgen biosynthetic defects, and incomplete gonadal dysgenesis, there is dissatisfaction with assigned genders in 25% of individuals whether raised as a boy or a girl [37]. The decision of sex of rearing in patients with ovotesticular DSDs must take into account the potential for fertility based on the degree of gonadal differentiation and genital development. Factors to consider for mixed gonadal dysgenesis also include prenatal androgen exposure, testicular function, phallic development, and gonadal location [4]. The general recommendations are to raise infants with 46,XX CAH or 46,XY CAIS as females, whereas for infants diagnosed with 5α-reductase deficiency or 17β-hydroxysteroid dehydrogenase- 3 deficiency, a male assignment should be considered [38].
SURGICAL MANAGEMENT OF AMBIGUOUS GENITALIA
1. The aim of surgery
Surgery can relieve parental distress and improve attachment between the child with DSD and the family [39-42]. The aim of surgery is to make ambiguous external genitalia compatible with assigned gender, preventing urinary obstruction or infections, preserving sexual and reproductive potentials, and maximizing anatomy to enhance sexual function [12]. Surgical corrections usually concern the gonads and the outer genitalia and often the presence of a urogenital sinus. There is no evidence that prophylactic removal of asymptomatic discordant structures is required. In general, it is recommended that the decision about genital surgery should be made by the parents and, when possible, the patient, under the counseling of the medical team. It is important to inform the parents that a functional outcome is more important than a cosmetic outcome. Patients and parents should not be given unrealistic expectations about penile reconstruction.
2. The timing of surgery
In the opinion of some experts, genital surgery in infancy that makes an appearance consistent with the gender of rearing is of significant psychological support to the family. However, others suggest that appearance-altering operation is not urgent and that it is more appropriate to delay surgery until a patient is old enough to be informed fully and to provide consent. Although there are still controversies about the optimal timing of the surgery, the American Academy of Pediatrics guidelines on the timing of genital surgery recommend genitoplasty between 2 and 6 months of age [43] and many pediatric urologists also recommend early feminizing genitoplasty [12,44]. Also, some studies have demonstrated satisfactory outcomes from early surgery [12,37,44,45]. The rationale for early reconstruction is based on the aforementioned guidelines on the timing of genital surgery from the American Academy of Pediatrics, the beneficial effects of estrogen in early time, preventing potentially harmful effects from the communication between the urinary tract and peritoneum through the fallopian tubes, minimizing family concerns, and alleviating the risks of stigmatization and gender-identity confusion [39,43,44].
3. Surgical procedures
Feminizing genitoplasty for the infants who are to be raised as females includes 1) removing the corporal bodies, 2) creating a normal-looking introitus and labia minora and majora, and 3) vaginoplasty to provide an adequate opening. Masculine reconstruction may include 1) orchiopexy, 2) hypospadias repair, and 3) removal of retained müllerian duct structures.
4. Clitoroplasty
Clitoroplasty should be considered only in cases of severe virilization and should be performed combined with repair of the common urogenital sinus. Until the 1960s, the principal surgical procedure for clitoromegaly was clitoridectomy. Amputation of the clitoris leaves a female-appearing perineum but orgasmic function and erectile sensation may be disturbed. Total removal of the clitoris is contraindicated, and when clitoral reduction is performed, sparing the neurovascular bundle is important for the preservations of intact orgasmic function and erectile sensation [46,47].
5. Vaginoplasty
Some prefer to correct the external genitalia in a single-stage procedure in the newborn period to take advantage of all native genital tissue and to avoid scarring [40,48,49]. Others advocate postponement with the vaginal reconstruction until puberty when vaginal dilatations are more feasible to prevent vaginal stenosis [50]. Because the risk of vaginal stenosis is high following vaginoplasty before puberty, delayed vaginoplasty could be considered if the urinary drainage is adequate. Several options such as self-dilatation, skin substitution, and bowel vaginoplasty have specific advantages and disadvantages; thereby, no one technique could be invariably applied. To prevent stenosis, vaginal dilatation can begin 2 weeks after the operation. However, vaginal dilatation in early childhood is prohibited.
7. Phalloplasty
Currently, there is no tissue available to increase the size of an underdeveloped phallus in humans. In patients with DSD associated with hypospadias requiring phalloplasty, the complexity of this procedure must be discussed during the initial counseling and standard surgical repair including chordee correction, urethral reconstruction, and judicious testosterone supplementation is recommended [54].
8. Gonadectomy
Although there is controversy regarding the timing of gonadectomy, it is commonly recommended soon after diagnosis because estrogen-replacement therapy could be started [4,55]. The advantages of late gonadectomy include breast development and avoidance of poor adolescent compliance with estrogen-replacement therapy. Germ cell malignancy only occurs in patients with DSD who have Y-chromosomal material (GBY region). Testes in patients with 46,XY gonadal dysgenesis who are raised as a female should be removed to prevent testicular malignancy [55]. In patients with androgen biosynthetic defects raised female, gonadectomy should be performed before puberty. A scrotal testis in patients with gonadal dysgenesis is at risk for malignancy. Thereby, current recommendations are to perform testicular biopsy at puberty, seeking carcinoma in situ or undifferentiated intratubular germ cell neoplasia, which are premalignant lesions [56]. If positive, the option is sperm banking before treatment with local low-dose radiotherapy [56]. In female patients with ovotesticular DSD, the tumor risk is low (<5%), but removal of the testicular component in early life is recommended to preserve fertility potential [57].
STEROID REPLACEMENT
Hypogonadism is common in patients with dysgenetic gonads, defects in sex-steroid biosynthesis, and resistance to androgens. Hormone-replacement therapy is often required to induce and sustain puberty, induce secondary sexual characteristics and pubertal growth spurt, optimize bone mineral accumulation, and for psychosocial maturation in patients with DSD [58]. Boys with hypogonadism require intramuscular injections of either testosterone cypionate or ethanate for pubertal induction [59]. Other testosterone preparations such as gels and patches are available [60-62]. In girls with hypogonadism, estrogen supplementation to induce secondary sexual changes and menstruations is required. Estrogen can be given orally, by injection, orpatch [59]. A progestin is usually added after breakthrough bleeding develops or within 1 to 2 years of continuous estrogen.
PSYCHOSOCIAL SUPPORT
Current recommendations emphasize sensitive, supportive interactions with families, and full disclosures of the risks, benefits, and potential outcomes of intervention to allow them to participate as fully as possible in decision making and in the continuing care of their child. Psychosocial care should be an integral part of management to promote positive adaptation. Families and individuals require ongoing counseling by experienced personnel to identify maladaptive coping strategies, deal with challenges such as gender dysphoria and sexual dysfunction as they arise, and facilitate age-appropriate information sharing with their affected child. Other important resources include access to confidential sexual counseling and support groups. Regular follow-up from infancy to adulthood of sexual, psychological, and social parameters is needed to provide a favorable long-term outcome for patients with DSD.
CONCLUSIONS
DSD with ambiguous genitalia is a rare disorder requiring prompt investigation and early gender assignment that is logically based on a sound knowledge of normal sex determination and differentiation. Despite the significant advances that have been achieved, much remains to be clarified in terms of the accurate evaluation and optimal management of patients with DSD. The complexity of the problem requires a multidisciplinary team working together, including a pediatric urologist, pediatric endocrinologist, psychiatrist, and social workers for the proper management of patients with DSD. Affected patients and the parents should be provided with full information to make an appropriate choice for gender assignment. The aims of management in a newborn with DSD should be the provision of a stable gender identity with psychological support to the family, potential sexual function and fertility, and affirmative body image. The surgical correction technique and the timing of the operation need to be individualized according to medical conditions, experience of the surgeon, and the complexity of each case. The general trend is towards early reconstruction with subsequent early and long-term management of the patient. The discussion of the evaluation and management of patients with DSD continues. Future study with further data is essential to suggest enhanced clinical guidelines and recommendations fitting the case of each individual patient.


 XML Download
XML Download