

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Sarcomatoid urothelial carcinoma of the renal pelvis is an uncommon variant of urothelial carcinoma that has both malignant epithelial and mesenchymal components.1 This is a rare malignant tumor. Only 1 case of primary sarcomatoid urothelial carcinoma of the renal pelvis has been reported in Korea.2 This tumor is very difficult to diagnose, and immunohistochemistry is helpful for a correct diagnosis. The clinical behavior of this tumor is aggressive.3 We experienced a case of a sarcomatoid urothelial carcinoma of the renal pelvis with extremely aggressive clinical behavior in an 81-year-old woman.
CASE REPORT
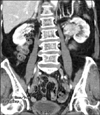
An 81-year-old woman presented with a 2 week history of painless gross hematuria. She was healthy with no medical problems and denied smoking and drug abuse. No mass was palpable in the abdomen during a physical examination. Urine cytology was suspicious of transitional cell carcinoma. Abdominal ultrasonography demonstrated dilation of the pelvocaliceal system of the left kidney. In the cystoscopic examination, jetting of hematuria from the left ureteral orifice was observed. Computed tomography (CT) revealed a 4.5×3.1 cm well-enhancing mass in the left renal pelvis and no distant metastasis (Fig. 1). Metastatic evaluation including chest CT and nuclear medicine bone scan demonstrated no sign of disease spread.
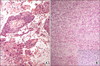
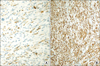
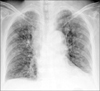
The patient underwent a left laparoscopic radical nephroureterectomy with no intraoperative problems. The entrapped specimen was divided into 3 pieces and removed through a 3 cm sized initial port site. Grossly, there was a 4.5×3×2 cm sized exophytic, multilobulated and polypoid mass at the renal pelvis. Histologically, the tumor consisted of neoplastic spindle cells that partly formed a fascicular arrangement with frequent vascular invasion (Fig. 2). Meticulous sections of the tumor revealed a few foci of urothelial components and merging of the epithelial and spindle cell components (Fig. 2). There was no heterologous element in the meticulous sections from the tumor mass. Immunohistochemical analysis was performed with antibodies against cytokeratin, smooth muscle-specific actin, desmin, and vimentin. The spindle cell elements showed coexpression of both cytokeratin and vimentin and were negative for muscle-specific actin and desmin (Fig. 3). These findings led to the diagnosis of a sarcomatoid urothelial carcinoma without heterologous elements according to the 2004 WHO classification of tumors of the urinary system and male genital organs.4 The postoperative period was unremarkable, and the patient was discharged home 5 days after the operation. One month after surgery, however, the patient was readmitted because of nausea, poor oral intake, and general weakness. Chest radiography showed multiple small nodules suspicious of a metastatic tumor in the whole lung field (Fig. 4). After admission, decreased anal tone and fecal incontinence suspicious of spinal cord compression occurred. The patient's general condition changed for the worse rapidly, and additional treatment could not be done. She died 2 months after surgery. An autopsy was not performed because her family did not provide consent.
DISCUSSION
Sarcomatoid urothelial carcinoma of the renal pelvis is a very unusual malignant tumor, consisting of an admixture of carcinomatous and sarcomatoid components. Histologically, the epithelial component is urothelial, and in some cases the epithelial component cannot be recognized.5 In the current case, only a few areas of the multiple sections from the tumor included high-grade papillary urothelial carcinoma. Immunohistochemical studies are mandatory for the diagnosis to prove biphasic differentiation such as cytokeratin showing epithelial differentiation and vimentin showing mesenchymal differentiation.4,6 Coexpression of vimentin and cytokeratin was demonstrable in the present case. According to the 2004 WHO classification of tumors of the urinary system and male genital organs, heterologous elements including chondrosarcoma, osteosarcoma, or rhabdomyosarcoma express markers appropriate to the type of differentiation; these include S-100, muscle-specific actin, desmin, and myoglobulin.4 The immunohistochemistry results for all of these markers were negative in the present case; in addition, histologically, heterologous elements were absent in the meticulous sections from the tumor mass.
Various terms have been used for these neoplasms, including carcinosarcoma, sarcomatoid carcinoma, spindle cell carcinoma, and giant cell carcinoma.4 Some authors prefer to use sarcomatoid carcinoma for those cases without heterologous elements in the spindle cell component and carcinosarcoma for cases with heterologous elements.6 However, considerable disagreement regarding the nomenclature and histogenesis of the neoplasms remains. Regardless of their histological features, however, the clinical characteristics of these neoplasms, including patient age, sex, presentation, and outcome, are known to be similar.
Clinically, sarcomatoid urothelial carcinoma of the renal pelvis is known to be rapid in progression and has a poor prognosis. Most previously reported patients died within 2 years. The mean survival time was shorter than 9 months. There are no reports on the effects of radiation therapy and chemotherapy on this tumor. Therefore, no specific guidelines regarding adjuvant therapy have been described. However, Thiel et al3 reported that one patient appeared to have a sarcomatoid urothelial carcinoma confined to the renal pelvis without signs of metastatic spread. After laparoscopic nephroureterectomy without adjuvant therapy, he was without recurrence for more than 1 year. Therefore, a radical, extensive operation seems to be most important to treat this disease.
Our patient died 2 months after surgery due to rapid metastasis to the whole body. Petsch et al7 reported a similar case. In their case, an 82-year-old woman underwent total nephrectomy for a ureteral sarcomatoid carcinoma. However, she developed "explosive" metastasis and died 2 months after surgery.
The experience in this case emphasizes that urothelial carcinomas can show divergent differentiation with different therapeutic ramifications; hence, the sarcomatoid variant should be managed as a highly aggressive tumor with poor outcome requiring more aggressive therapy.
 XML Download
XML Download