

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Castleman disease, or angiofollicular lymph node hyperplasia, is a fairly rare benign tumor of lymphoid origin with unknown etiology.1 Castleman disease arises mostly in the mediastinum. Some cases of non-mediastinal origin have been reported, but perirenal tumors are extremely rare. Diagnostic imaging methods cannot differentiate Castleman disease from other diseases because of the lack of tumor-specific signs. Here we present a case of perirenal Castleman disease misdiagnosed as a neoplasm of the kidney, with a review of the literature.
CASE REPORT
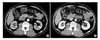
A 58-year-old male patient was incidentally found to have a right renal mass on abdominal ultrasonography (US) during a health inspection while living abroad. He denied having any symptoms including abdominal pain or discomfort or gross hematuria and looked healthy. He had hypertension and diabetes mellitus diagnosed 15 years ago and had been treated with medication. The physical examination showed no remarkable findings such as a palpable mass or costovertebral angle tenderness. Urinalysis, complete blood count, and routine blood biochemistry results were within normal limits and the results of a human immunodeficiency virus (HIV) antibody test were negative. Urine cytology showed no evidence of malignancy. Abdominal US demonstrated a 2.0×1.8 cm, well-demarcated, isoechoic mass at the medial cortex of the right kidney, and abdominal computed tomography (CT) also revealed a similar sized, well-defined, solid enhancing mass at the medial cortex of the right kidney (Fig. 1). Generally, with contrast-enhanced CT, conventional renal cell carcinomas (conventional RCCs) have dense enhancement at the arterial phase and washout to low density at the delayed phase, but the CT findings of our patient did not correspond with those of conventional RCC. Accordingly, we suspected the mass to be nonconventional RCC or some other type of renal tumor such as oncocytoma. We decided to operate because we could not completely rule out the possibility of malignancy. At first, we considered a partial nephrectomy, because the mass size was only 2 cm in diameter. But the location of the mass was around the sinusoidal area at the medial portion of the kidney and very close to renal vessels; therefore, we reasoned that partial nephrectomy would be difficult to perform because the lesion was not easy to access. The patient underwent radical nephrectomy through a right anterior subcostal incision with a transperitoneal approach under general anesthesia. There was no specific intraoperative finding.
Grossly, the specimen contained a 1.8×1.5×1.0 cm, grayish brown, ovoid mass in the hilar soft tissue. The mass abutted the renal cortex without necrosis (Fig. 2). Microscopically, the tumor had completely encapsulated lymph nodes. Hyperplastic follicles were shown in a mass of lymphoid tissue, and the interfollicular area contained sheets of plasma cells (Fig. 3). We recognized the presence of a polyclonal process by immunohistochemical stains for κ (kappa) and λ (lambda) chains. The stain for human herpes virus 8 (HHV-8) was negative. The remaining renal parenchyma and adrenal gland were unremarkable. These histologic findings were consistent with the plasma cell type of Castleman disease. Three months after the operation, CT of the chest and abdomen was performed for follow-up, and there was no sign of recurrence of the disease. The patient has remained in a good general condition without abnormal physical signs.
DISCUSSION
Castleman disease is a rare heterogeneous lymphoproliferative disorder of unknown etiology that was first described by Castleman in 1954.1 Castleman disease arises mostly in the mediastinum, and about 70% of the reported cases tend to be of mediastinum origin. The superficial nodal groups constitute 20% of cases. Retroperitoneal and pararenal localizations are very rare and have been reported to account for 7% and 2% of cases, respectively.2 Our patient had Castleman disease just around the kidney; most of the patients with these tumors present with an asymptomatic abdominal mass or with vague abdominal pain resulting from the mass effect. Rarely, a mass that compresses the renal pelvis or proximal ureter can cause hydronephrosis.3
Castleman disease is clinically classified into unicentric type and multicentric type and is histologically classified into hyaline vascular type, plasma cell type, and mixed type.
The unicentric hyaline vascular type generally affects young people and occurs in approximately 70% of the patients with Castleman disease.4 It is usually asymptomatic.2 Because of its vascularization, there is a risk of massive hemorrhage during surgery.
The unicentric plasma cell type occurs in approximately 20% of patients with Castleman disease. It is characterized by hypertrophy of a single lymph node chain.4 The majority of such patients present with constitutional symptoms. Anemia and an elevated erythrocyte sedimentation rate (ESR) are present in most cases.5 These findings did not correspond with those of our patient, who did not have abdominal pain or discomfort or abnormal laboratory results.
The multicentric plasma cell type is the least commonly encountered Castleman disease variant (10%), but presents with the most protean manifestations. Patients with this type are typically older than those with unicentric disease. The most common of systemic symptoms in this type are anemia, fever, diaphoresis, weight loss, night sweating, and fatigue.4 Patients with this type may have an elevated ESR, hyperglobulinemia, hypoalbuminemia, polyclonal hypergammaglobulinemia, leukocytosis, thrombocytosis, or splenomegaly.5,6 The deregulated overproduction of interleukin-6 is responsible for these findings.2,7
The plasma cell type can be found in autoimmune disease, AIDS, and lymph-node draining carcinoma, so it is imperative to exclude this condition before diagnosing the plasma cell type of Castleman disease.8 Especially, the multicentric plasma cell type is highly associated with infection by HHV-8, and patients have an increased risk of development of HHV-8-associated neoplasms, including Kaposi sarcoma and extranodal B-cell lymphoma.7
Diagnostic imaging methods, such as US, CT, and magnetic resonance imaging generally cannot differentiate Castleman disease from other diseases because of the lack of tumor-specific signs, but these examinations do enable us to locate the exact position of a tumor.2 We suggest that for tumors having characteristics of homogeneous soft tissue density and hypervascularity in areas where lymphoid tissue is normally found, the hyaline vascular type of Castleman disease should be added to the list of differential diagnosis.9 In our case, the preoperative radiologic findings matched with the hyaline vascular type because of the enhancing mass shown on CT, but the postoperative histological diagnosis was the plasma cell type. Even though renal biopsy can be undertaken to assess the malignancy of the tumor and to help to avoid extensive resection in the case of the absence of a preliminary diagnosis,8 most clinicians rarely perform biopsy of an enhancing renal mass.
In patients with lymph node proliferation consistent with Castleman disease, other causes of reactive lymphadenopathy that look similar should first be ruled out, such as rheumatoid arthritis, lupus, Sjögren syndrome, HIV infection, lymphoma, and drug sensitivity.5
Unicentric Castleman disease of either the hyaline vascular or plasma cell type is almost universally cured after resection of the lymph nodes involved and is not associated with increased mortality.4 Particularly for the unicentric hyaline vascular type, if complete resection is not feasible, partial resection alone can be expected to improve symptoms and has a high survival rate,10 and long-term observation can also be a possible choice.2 Radiotherapy may be a viable option for patients who are considered to be poor surgical candidates or in cases with an incomplete resection.2 Because of the disseminated nature of the lymphadenopathy seen in multicentric disease, complete surgical debulking is rarely possible,5 so the treatment method for the multicentric form has not yet been firmly established. Systemic therapy with steroids or antiblastic agents has been used with variable success.4 Ganciclovir, interferon-α, or rituximab may be the best treatment options for patients with HHV-8 infection, whereas cyclophosphamide, vincristine, doxorubicin, and either prednisone (CHOP) or dexamethasone (CVAD) may be the most appropriate for patients with severe systemic manifestations of multicentric Castleman disease.5 Recently, humanized anti-interleukin-6 receptor antibody was reported to be an effective treatment for multicentric Castleman disease, but the benefit of this has not been definitely proved yet.2,10
Patients with unicentric disease without systemic involvement should have an additional radiological assessment 6 to 12 months after therapy to verify the cure, with additional testing or therapy pursued only in the event of the onset of new symptoms.5 For patients with multicentric Castleman disease, close follow-up and periodic surveillance are necessary, so that concurrent or ensuing malignant lesions can be detected.4
The prognosis is poor for the plasma cell type, particularly in multicentric Castleman disease, because of the high incidence of malignancy.6 We expect that our patient will have a good prognosis because his disease was the unicentric type without constitutional symptoms and he was negative for HHV-8. But because of the plasma cell type, our patient will require periodic serum testing as well as radiologic study.
A preoperative diagnosis of Castleman disease is difficult; therefore, a surgical resection and a histological evaluation can provide an accurate diagnosis of this tumor. In consideration of the present case, we suggest that Castleman disease should be included in the differential diagnosis of renal tumors.


 XML Download
XML Download