

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Carcinoid tumors are malignant tumors derived from neuroendocrine enterochromaffin cells. Carcinoid tumors can occur on any organ but most frequently occur in digestive and respiratory organs. They have also been reported to arise in the breasts, ovaries, testicles, and gallbladder. On the other hand, they rarely occur in the organs of the retroperitoneum, because of the nonexistence of neuroendocrine cells in normal kidney, renal pelvis, and ureters. To date, approximately 50 cases of carcinoid tumors that have arisen in the kidneys have been reported in the literature, but no clear cause or prognosis for these tumors has been established. In Korea, a case of carcinoid tumor that occurred in a horseshoe kidney was reported.1 Here we present a case of a primary renal carcinoid tumor diagnosed after radical nephrectomy in a patient in whom the renal mass was found incidentally during a comprehensive medical examination.
CASE REPORT
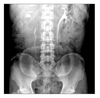
A 51-year-old male patient came to the hospital because of a tumor in his right kidney, which was accidentally discovered on abdomen ultrasonography during a comprehensive medical examination. The patient looked well in appearance and had no specific matter in his medical or family history. No mass was palpable and no tenderness was observed in the right upper quadrant. The results of a complete blood count, urine analysis, and routine chemistry tests were within the normal range, and there was no specific finding except a calcified lesion of the right upper quadrant on a plain frontal supine radiograph of the abdomen.
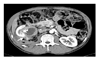
Excretory urography showed that the renal pelvis was compressed by the mass; by use of ultrasonography, the cystic mass was identified in the medial side of the right kidney with unclear boundaries to adjacent organs (Fig. 1). On the abdominal CT scan, the tumors seemed to be compressing the renal parenchyme laterally. The tumor appeared as a cystic mass about 7.0×5.2 cm in size containing many calcified tubers with necrosis interiorly and a relatively thick wall. Also, the tumor was suspected to be infiltrating the lymph nodes in the perirenal space and around the vena cava (Fig. 2).
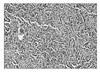
In these examinations, we suspected a malignant cystic renal cell carcinoma, and we then performed a transperitoneal radical nephrectomy. In the operating sight, the tumor was located between the medial side of the renal cortex and the vena cava and had severe adhesion with adjoined tissue. Lymph node enlargement of about 1.5 cm was observed between the right renal artery and the vena cava. The mass was observed as a cystic structure and hard tubercles just like bony fragment were stuck into the cystic mass, with a light yellowish abscess formed around it. At the biopsy after surgery, a well-differentiated neuroendocrine tumor was confirmed. The tissues were in the form of a typical carcinoid tumor containing eosinophilic infiltration conjoined with small, monotonous, cuboidal cells. No mitosis was seen (Fig. 3). Among the 7 excised lymph nodes, 6 lymph nodes had a carcinoid texture, including the right hilar lymph nodes. NSE and synaptophysin stained positive by immunohistochemical staining.
In the retrospective research carried out after the surgery, the patient did not show symptoms of carcinoid syndromes, such as facial hot flushing and diarrhea. On an 111Indium-labelled octreotide scan, which was done 1 month after surgery, no relatively hot uptake lesions were seen. Thus, we diagnosed the tumor as a primary renal carcinoid tumor without distant metastasis, and we did not perform additional chemotherapy or radiation therapy. The patient has not shown any sign of recurrence for 3 months after surgery while followed up as an outpatient.
DISCUSSION
Carcinoid tumors are malignant tumors derived from neuroendocrine enterochromaffin cells that can occur in any organ. They develop most often in the digestive organs (84%%), including the small intestines, and the respiratory organs (10%) and may seldom arise in the kidney, breast, ovaries, and prostate.2 As for the cause of kidney carcinoid tumors, it is hypothesized that they come from the postnatal urothelial metaplasia related to chronic inflammation and that they develop congenitally by errors in location choice of neural crest or pancreas tissues during the renal embryogenic process. However, none of these hypotheses has been clearly proved.3-5
Carcinoid tumors occurring in the kidney often develop throughout middle age (mean age, 49 years), and are known to differ little by sex or laterality. Renal carcinoid tumors can cause either localized symptoms, such as hematuria, abdominal pain, or flank pain, or carcinoid syndrome, that is, systemic symptoms, such as perspiration and diarrhea. However, cases without any symptoms are often reported. Shurtleff et al6 reviewed the literature of 43 cases of primary kidney carcinoid tumors and reported that abdominal pain and flank pain with or without hematuria was the most frequent clinical symptom (55.8%). Six (13.6%) cases showed carcinoid syndrome, and 9 (20.5%) were diagnosed as renal masses found accidentally during imaging studies of patients with no clinical symptoms. Romero et al7 reported in research carried out on 56 cases of renal carcinoid tumors that 26.8% were accompanied by other pathologic lesions inside the kidneys, including calcification within tumors and cystic change, and only 7% showed carcinoid syndromes that presented systemic symptoms.
Because carcinoid tumors do not show endocrinologic symptoms, it is difficult to diagnose and predict the existence of these tumors by the results of blood tests. The trial of serum analysis measuring the quantity of serotonin, histamine, prostaglandin, and bradykinin is ongoing, but the effectiveness of this has not been established yet. Carcinoid tumors are generally diagnosed through biopsy after surgery, because the benefits of radiologic examinations have not been proved. Carcinoid tumors resemble in appearance typical tumors inside the kidney, such as renal cell carcinoma. An abdominal CT scan is known to be the most useful tool for finding the mass and setting up the stage. About 26.5% of the imaging studies showed accompanied calcified lesions interior to the mass, and at the time of diagnosis, about half of the lymph nodes around the aorta or near the renal hilum showed invasion.7 Distant metastasis was most common in the liver (34%).7 Mckeown et al8 reported that the renal carcinoid tumors appeared as an atypical renal cell carcinoma in 3 cases of primary renal carcinoid tumors among 10, and the others appeared as renal masses, which are hard to distinguish clearly from renal cell carcinoma. In our patient, the mass was seen as an atypical renal cell carcinoma, which is accompanied by internal calcification.
For renal carcinoid tumors, the best treatment is complete surgical resection of the tumor. Generally, radical nephrectomy with adjacent lymph node dissection is the treatment of choice for renal carcinoid tumors; if the mass is small and confined to the kidney, partial nephrectomy may be possible. In the case of invasion to organs other than the liver, systemic chemotherapy can be tried, but the effect is known to be feeble on carcinoid tumors in digestive or respiratory systems.9 In the case of metastasis to the liver with a single segment or lobe, embolization or chemoembolization can be performed for the metastatic nodule in the liver. Recently, interferon -α, -γ, or somatostatin analogue, which are used for carcinoid syndrome of the digestive organ, are being tried, but the benefits of this therapy have not been established yet.10
Clinical prognosis is known to differ according to the region in which the tumors arise, the size of the tumors, and the general conditions of the patient. In general, the course turns out to be better than for renal cell carcinomas, and the prognosis is known to be comparably good if complete excision of the tumor can be done. If the mass is more than 4 cm in size and local lymph node invasion is present, distance metastasis is more common than not. Shurtleff et al6 observed 43 patients for a mean follow-up time of 27.3 months and reported that 4 cases relapsed after the surgery (9%) and 4 died from the tumor (9%).
Primary renal carcinoid tumors with such traits have no clear clinical progress or indicator for clinical follow-up yet, for there are few reported examples, no large unit research, and no data from long-term surveys. Constant investigation and accumulation of cases of such tumors are necessary.
 XML Download
XML Download