

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
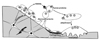
In the adult human skeleton, approximately 5 to 10% of the existing bone is replaced every year by bone remodelling. The remodelling process, which continues throughout adult life, provides for the calcium homeostatic system and is essential for resorptive removal of old bone, the removal and repair of micro-damage, and for the adaptation to mechanical stress[1,2]. The cellular sequence is initiated with signals that lead to osteoclast development and bone resorption (Fig. 1). How those signals are initiated is uncertain. In the case of micro-damage, this is proposed to lead to apoptosis of osteocytes that transmit signals to surface cells to promote production of receptor activator of NFκB ligand (RANKL) and hence osteoclast production[3].
Remodelling is essential for the maintenance of skeletal material and structural strength, with bone being continuously resorbed and reformed at about 1~2 million microscopic remodelling foci per adult skeleton. This sequence of events is initiated asynchronously throughout the skeleton, at sites that are geographically and chronologically separated from each other. Both bone resorption and bone formation occur at the same place in these "basic multicellular units" (BMUs), so that there is no change in the shape of the bone[4]. Within each of these BMUs, focal resorption is carried out by haemopoietically-derived osteoclasts and takes about 3 weeks per site, whereas the refilling of lost bone by osteoblasts, derived from bone marrow stromal cells and circulating precursors, takes about 3~4 months.
In addition to remodelling, bone modelling on its periosteal surface is characterised by bone formation without prior bone resorption. This process, so vigorous during growth, establishes the adult size and shape of bone. At the completion of linear growth with closure of the epiphyses, periosteal apposition continues but markedly less so[5]. Tight regulation of these processes is essential for the achievement and maintenance of skeletal strength. Modelling and remodelling during growth achieves peak bone strength, and continued remodelling during adulthood maintains the mechanical integrity of the skeleton. Circulating hormones contribute, but the key influences are locally generated cytokines that are the signals mediating information transfer among osteoblasts, osteoclasts, immune cells and constituents of the bone matrix.
Coupling of Bone Formation to Resorption
The maintenance of bone mass in accordance with loading requirements requires that bone resorption and formation in remodelling be as near as possible to equal, so that a high or low level of resorption is associated with a similar change in focal bone formation in the BMU. The theory that resorption is followed by an equal amount of formation is crucial, and has come to be known as "coupling"[1]. When coupling is disturbed so that resorption exceeds formation in the BMU, bone loss follows. Thus uncoupling occurs with estrogen withdrawal, weightlessness, ageing, and cancer effects on bone (e.g. especially multiple myeloma). On the other hand a positive balance, with formation exceeding resorption, is the case during growth.
A striking example of the dissociation of coupling of bone formation to resorption is that resulting from estrogen removal following ovariectomy or menopause. Attempts have been made to explain the increased resorption and loss of bone with estrogen withdrawal, by increased production and/or activity of any of several cytokines - IL-6, IL-1, TNFα, RANKL[6~8], with the common mechanism being a deficiency in the coupling process. Drugs such as bisphosphonates and SERMs, used in the treatment of osteoporosis, reduce the excessive resorption, but this is accompanied by reduced bone formation because of the coupling of the two processes. This limits the benefits that can be obtained from their use, and most importantly, on current evidence[2,9] argues against their combined use with the only available bone anabolic treatment, parathyroid hormone (PTH) given by daily subcutaneous injection[10,11]. On the other hand, a resorption inhibitor that does not inhibit bone formation would be ideal to combine with anabolic therapy. Identification of coupling factor would reveal a locally generated molecule that is an important regulator of the rate and extent of bone formation, and therefore one which could provide valuable approaches to enhancing existing treatments of bone disease by dissociating inhibition of bone formation from inhibition of formation. At the present time there are few experimental models in which coupling is dissociated. In the delta FosB-overexpressing transgenic mice bone formation is increased without any increase in resorption[12]. In mice deficient in either c-src[13] or the cathepsin K[14], resorption is inhibited without inhibition of formation. In each of these two knockout mouse lines osteoclast resorption is greatly reduced by the mutation, although osteoclast numbers are not reduced.
Osteoclastic resorption takes place in a sealed-off microenvironment, whereby the osteoclasts attach surrounded by a peripheral ring tightly adherent to the bone matrix, thus sealing off the sub-osteoclastic resorbing compartment. The necessary acidification of this compartment results from the H+ and accompanying Cl- ions produced by the cells, with the acid pH favouring dissolution of the bone mineral, thereby exposing the organic matrix to proteolytic enzymes. These enzymes, which include collagenases and cathepsin K, are responsible for the degradation of the organic matrix.
Coupling Factor or Several Contributors to the Process?
A local "coupling factor" linking bone resorption to subsequent formation has long been proposed as the key regulator of the remodelling process, but never identified. Although there have been suggestions that such coupling activity is released from the bone matrix during resorption, there is now much interest in the possibility that coupling activity emanates from the osteoclast contributing to the ability of osteoblast lineage cells to differentiate to form bone and replace the resorbed bone in each BMU. Osteoclasts and the osteoblasts are intimately linked in several ways we proposed in 1981[15], that osteoblastic lineage cells mediate hormonal stimulation of osteoclast formation and activity, and ultimately the TNF family member, RANKL, was identified as the membrane protein expressed by the osteoblast lineage that is primarily responsible for promoting osteoclast formation and activity. Now it is necessary to examine how osteoclasts communicate with osteoblasts in the opposite direction.
A coupling mechanism linking bone resorption to subsequent formation was proposed by Howard and colleagues as the key regulator of the remodelling process[16]. The concept developed that coupling might be achieved by the activities of one or more growth factors released from bone matrix during resorption, with most credence given to IGF-I and II, and TGFβ[17~19]. These growth factors are stored in large amounts in bone, and the theory proposes that upon resorption of matrix they are activated by the prevailing acid pH, thus becoming available to act upon osteoblast precursors to promote bone formation. One specific function ascribed to TGFβ-1 is that upon its release with bone resorption, it can induce bone marrow stromal cell migration to resorbed surfaces, making them available for osteoblast differentiation under the influence of any or all of other growth factors and cytokines that become available in that environment[20]. Such a mechanism might go some way towards providing for the fact that local osteoblast precursors are many microns away from sites of osteoclastic resorption. It seems unlikely, though, that this would be an exclusive mechanism of providing osteoblast precursors to a BMU, since they might arrive via the circulation through capillaries penetrating the canopy (bone remodeling compartment (BRC)) that overlies the BMU[21~23].
Several questions should be considered regarding the role of TGFβ and other growth factors in the coupling of bone formation to bone resorption: (i) which cells produce them and under what circumstances; (ii) do they stimulate bone formation in vivo; (iii) can they be released from the matrix in active form and in controlled amounts during bone resorption; (iv) is there evidence for an increase in the abundance of these substances at sites of bone remodeling and (v) are there regulated mechanisms by which they are activated. The latter is most important, relating to the time course and the distance between the resorption and formation processes, and whether the amount of growth factors, and therefore the extent of activation of the osteoblast lineage, can be controlled with sufficient precision in this way.
Clearly there are many steps required to achieve precision in the coupling process: precursor replication, differentiation, limiting the cell mass to exactly what is required, and the correct shaping. It seems unlikely that any single factor dominates in control of the coupling process.
Could the Osteoclast Produce Coupling Activity?
Some recent studies in genetically manipulated mice provide indications that the osteoclast itself could be the source of an activity that contributes to the fine control that is a feature of the coupling process. The cytokines that signal through gp130 play an important role in intercellular communication processes in bone[24]. In studying mice in which each of the two gp130-dependent signalling pathways was specifically attenuated, we found that inactivation of the SHP2/ras/MAPK signalling pathway (gp130Y757F/Y757F mice) yielded mice with greater osteoclast numbers and bone resorption, as well as greater bone formation than wild type mice. This increased bone remodelling resulted in less bone because the increase in resorption was relatively greater than that in formation. In other words the coupling process was dissociated in a way that resembles the result of estrogen withdrawal, as in ovariectomy. gp130Y757F/Y757F mice crossed with IL-6 null mice had similarly high osteoclast numbers and increased bone resorption, however these mice showed no corresponding increase in bone formation and thus had extremely low bone mass. Thus resorption alone is insufficient to maintain the increased bone formation, but the active osteoclasts are the likely source. Furthermore this indicated that stimulation of bone formation coupled to the high level of bone resorption in gp130Y757F/Y757F mice is an IL-6-dependent process, though it does not necessarily show that it is mediated by IL-6 itself. This 1L-6-dependent mechanism is likely emanating from the active osteoclasts[25].
In osteopetrosis the amount of bone is increased because of the failure of bone resorption, either because osteoclasts are absent or inactive. In most forms of this condition the reduced resorption results in decreased bone formation because of the coupling of the processes. For example mice lacking c-fos, which are unable to generate osteoclasts[26], have reduced bone formation as well as resorption. In mice deficient in either c-src[13] or cathepsin K[14], however, bone resorption is inhibited without inhibition of the rate and extent of formation. Similarly, in human subjects with autosomal dominant osteopetrosis type II due to inactivating mutations of ClC-7, bone resorption is inhibited without decrease in the rate of bone formation[27]. A possibility in all these situations is that the osteoclasts are capable of generating a factor (or factors) required for bone formation, despite the fact that they do not resorb bone because of failure to acidify the resorption space.
In reviewing these studies with genetically manipulated mice and earlier evidence, it seems likely that there are many contributors to the overall process that results in the coupling of bone formation to resorption. These include particularly the release of growth factors from matrix, as well as the control from osteoclast-derived products. The control of the process is so precise however, that regulated activity, derived from the osteoclast, could work co-operatively with other growth factors to exert a powerful influence, most likely at an early stage in the process to influence the commitment of precursors within the osteoblast lineage.
How Do Cells of the Osteoblast Lineage Know How Much Bone to Make in a BMU?
Among the important unanswered questions concerning bone remodelling is why osteoclasts stop resorbing after excavating a certain amount of bone, and either die or move on. It is equally important to understand how the osteoblast lineage cells, forming bone within the BMU, replace virtually precisely the amount of bone that has been lost.
The next major stage of the remodelling process is the recruitment and differentiation of mesenchymal precursors to osteoblasts, sufficient to synthesize the amount of bone lost in the resorption process at that site. Of the several potential sources of osteoblast precursors, one is that lining cells, the single layer of flattened cells that have ceased their bone-forming function, can revert to that activity. Other likely sources are adjacent marrow stromal cell precursors and blood-borne osteoblast precursors that could be presented to the BMU from capillaries within the sinus structure provided by the BRC underneath its canopy[21,28]. Osteoblast progenitors are associated with vascular structures in the marrow and several studies suggest there may also be common progenitors giving rise to cells forming the blood vessel and pluripotent perivascular cells[29~33].
An interesting possibility comes from the work of Gray et al.[34], who showed in vitro that if they provided rat calvarial cells to bone slices with crevices and grooves excavated on them, the cells made bone in those defects, filling them exactly to a flat surface. The findings suggested that the topography of the bone affected the timing, siting and extent of new bone formation, and that in vivo this would take place in the resorbed spaces vacated by osteoclasts, with the participating cells themselves able to sense the spatial limits, and most likely doing so by autocrine and paracrine chemical communication. Growth factors and cytokines produced by the osteoblasts are candidate mediators of this process, as are gap junction communication between the osteoblasts themselves[35]. Regulation from outside the BMU population could be provided by osteoblast lineage cells residing within the bone matrix (osteocytes) that respond to mechanical changes in pressure and communicate with surface cells. Osteocytes produce sclerostin, which is a powerful inhibitor of bone formation that is suppressed by several stimuli that increase bone mass.
Another possible local control mechanism comes with the finding that PTH and PTHrP promote production by osteoblasts of ephrinB2 which acts through its receptor, EphB4, to promote osteoblast differentiation and bone formation within the BMU[36]. Ephrin/Eph family members are recognised as local mediators of cell function through contact-dependent processes in development and in maturity[37,38]. A particular feature is their capacity for bi-directional signalling, in that when an ephrin acts upon its corresponding Eph receptor tyrosine kinase, the latter can signal in the reverse direction, acting on ligand by promoting rapid phosphorylation on highly conserved tyrosine residues within the PDZ domain[39,40]. The main ligand for EphB4 is ephrinB2, with ephrinB1 interacting with much less affinity[40]. Some evidence suggests that osteoclast-derived ephrinB2 acts through a contact-dependent mechanism on EphB4 in osteoblasts, to promote osteoblast differentiation and bone formation, and that through reverse signalling, osteoblast-derived EphB4 acts upon ephrinB2 in osteoclasts to suppress osteoclast formation[41]. The latter mechanism would require osteoblast-osteoclast contact, with evidence for the proximity of EphB4-positive osteoblast precursors to osteoclasts being provided by Luiz de Freitas et al.[42]. In the same work ephrinB2-positive osteoblasts were close to bone formation sites, with the overall conclusion that the ephrin/Eph signalling pathway could function both within the osteoblast lineage to favour formation of bone[36], and between osteoblast and osteoclast to limit resorption[41]. These mechanisms warrant further study since pharmacological manipulation of ephrinB2/EphB4 signaling might be an attractive means of regulating the volumes of bone formed in the BMU.
Communication from the Osteocyte
Among the newly recognised functions of the osteocyte, its role in limiting bone formation is having a major impact on thinking in bone biology. Mutations in the SOST gene were found responsible for the rare sclerosing bone dysplasias, sclerosteosis and Van Buchem disease. Each is characterized by a greatly increased amount of bone. The SOST gene product, sclerostin, is produced exclusively in bone by osteocytes, and is a negative regulator of bone formation, inhibiting Wnt signalling through binding to the LRP5/6, thus preventing its participation in the receptor complex that activates the Wnt pathway and bone formation[43]. Production of sclerostin by osteocytes is rapidly decreased by treatment with PTH or PTHrP[44,45], as well as by mechanical loading[46]. For each of these stimuli, removal of sclerostin as a constitutive inhibitor of bone formation could at least partly explain the accompanying increased bone formation. Physiologically, rapid changes in osteocyte production of sclerostin could signal to surface cells to limit the filling of remodeling spaces by osteoblasts, in addition to keeping lining cells in a quiescent state on non-remodeling bone surfaces.
A further controlling influence upon osteoblast differentiation from within the osteoblast lineage comes from a study that identified a mechanism in which mature osteoblasts directed the differentiation of early mesenchymal osteoblast precursors through activation of canonical Wnt signalling[47]. Such a mechanism has the added advantage that its regulation would be susceptible to inhibition by osteoblast-derived inhibitors of Wnt signaling, such as a dickopf (Dkk) protein or secreted frizzled related protein (sFRP). Zhou et al.[47] provided evidence for inhibition of the process by sFRP1.
Conclusions
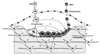
The idea of coupling of bone formation to resorption during remodelling has been much discussed since the early 1980's, with the thought being over many years that a single "coupling factor" might be responsible for this. With increasing understanding of the communication processes that take place among the cells of bone however, it is becoming clear that many pathways contribute to the coupling process, as depicted in the cartoon of Figure 2. Much is to be learned from studying each of these pathways, and disturbing any one of them can perturb the delicate balance that results in the tight control of bone remodelling. The study of these pathways has much to offer to our understanding of physiology and pathology, and progress towards developing pharmacological interventions that can influence bone remodelling.
 XML Download
XML Download