

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Multiple endocrine neoplasia type 2A (MEN 2A) is an autosomal dominant syndrome characterized by medullary thyroid carcinoma, pheochromocytoma and multigland parathyroid tumors. Sipple first described the syndrome in 1961 and germline activating mutations in the RET proto-oncogene have been identified as the disease-causing mutation involved in these inherited syndromes. A specific RET codon mutation is associated with the MEN 2 syndrome variant, the age at onset of the medullary thyroid carcinoma (MTC) and the aggressiveness of the MTC[1]. In Korea, Lee et al[2] first reported mutations of RET proto-oncogene in three Korean families with MEN 2A in 1998. The ectopic adrenocorticotropic hormone (ACTH) syndrome is characterized by hypercortisolism due to the hypersecretion of ACTH outside of the pituitary gland, which leads to Cushing's syndrome. The most common causes are malignancies such as a small cell carcinoma of the bronchus only rarely has an adrenal pheochromocytoma been identified as the cause[3,4].
Recently we treated a 29-year-old man with the ectopic ACTH syndrome with bilateral pheochromocytoma that had multiple endocrine neoplasia type 2A. The patient presented with medullary thyroid carcinoma and hyperparathyroidism. This is the first confirmed case of ectopic ACTH syndrome with bilateral pheochromocytoma in a Korean patient with multiple endocrine neoplasia type 2A.
Case
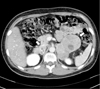
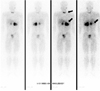
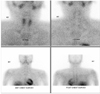
A 29-year-old man presented with a 1-year history of headache, hypertension, and exacerbated hyperglycemia. The physical examination showed a height of 167 cm and weight of 75.9 kg (body mass index 27.21 kg/m2). The systolic/diastolic blood pressure was 130 to 150/80 to 100 mmHg and the heart rate was 88 to 94 beats/min with medications (amlodipine 5 mg, ramipril/hydrochlorothiazide 2.5/12/5 mg). The abdominal computed tomography revealed a 10 cm heterogeneous enhancing mass with a lobulated contour on the left adrenal gland, and two focal 2.5 cm enhancing masses on the right adrenal gland (Fig. 1). Laboratory examinations revealed a leukocytosis (white blood cell count 14,900/µL), hyperglycemia (552 mg/dL) and mild hypercalcemia (total calcium 10.8 mg/dL, ionized calcium 4.64 mg/dL). Serum phosphorus level was in normal range (2.8 mg/dL). Endocrine profiles showed increased levels of 4 PM plasma ACTH (32.5 pg/mL; normal at 8 AM, 9 to 52 pg/mL, Radioimmunoassay Brahms ACTH RIA, Henningsdorf, Germany) and cortisol (44.5 µg/dL; normal 8 to 25 µg/dL). Overnight dexamethasone (1 mg) failed to suppress the endogenous cortisol secretion. Neither low dose dexamethasone nor high dose dexamethasone evoked any change in the plasma ACTH levels. These findings suggested autonomic secretion of ACTH and cortisol, although the patient had no typical Cushing features. The results of the tests of adrenal function are shown in Table 1. There was an elevated plasma calcitonin level 116.40 pg/mL (normal, ≤ 10 pg/mL) and elevated intact PTH level 158.0 pg/mL (normal, 8 to 76 pg/mL). Urinary hormone levels were as follows: free-cortisol 75.6 µg/day (normal, 20 to 90 µg/day), 17-OHCS 4.43 mg/day (normal, 3.6 to 9 mg/day), epinephrine 1060.0 µg/day (normal, ≤ 20 µg/day), norepinephrine 1464.1 µg/day (normal, 15 to 80 µg/day), dopamine 352.4 µg/day (normal, 65 to 400 µg/day), vanillylmandelic acid (VMA) 31.82 mg/day (normal, 0 to 8 mg/d), metanephrine 36.241 mg/d (normal, ≤ 1.3 mg/d), and normetanephrine 0.15 mg/d (normal, 0.07 to 0.26 mg/d). The 131-Iodine metaiodobenzylguanidine (MIBG) scintigraphy revealed high uptake in both adrenal glands (Fig. 2). Further ultrasound examination revealed two hypoechoic nodules on the right side of the thyroid and one hypoechoic nodule on the left. Fine needle aspiration of the nodules confirmed medullary thyroid carcinoma. The F-18 FDG PET CT showed a focal hypermetabolic lesion on the left side of the thyroid gland suggesting malignancies, a less intense hypermetabolic lesion on the right side of the thyroid gland and heterogeneous hypermetabolic masses on both adrenal glands (Fig. 3). The Tc-99m Pertechnetate sestamibi parathyroid scan showed no residual uptake (Fig. 4).
Informed consent for DNA analysis was obtained from the patient. Genomic DNA was isolated from peripheral blood leukocytes using the Wizard Genomic DNA Purification kit according to the manufacturer's instructions (Promega, Madison, WI, USA). We performed a polymerase chain reaction (PCR) and direct sequencing of Exons 10, 11 and 13~16 and their flanking sequences of the RET gene using primer pairs. PCR was performed in a thermal cycler (Model 9600, Applied Biosystems, Foster City, CA, USA), and cycle sequencing was performed in the ABI Prism 3130 Genetic Analyzer with the BigDye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems). Sequence variations were analyzed with reference to the wild type sequence using the Sequencher program (Gene Codes Corp., Ann Arbor, MI, USA). Mutational analysis of the RET gene detected a T1900 to C transversion, which led to a C634R amino acid change in exon 11 of the RET gene.
On admission, medication with insulin glargine (20~58 U/day) was started for hyperglycemia. Hypertension and tachycardia were treated with an alpha1-adrenergic antagonist (doxazosin mesilate) 8 mg/day. Two weeks after administration of the alpha1-adrenergic antagonist, elective surgery was planned. Both open adrenalectomy and total thyroidectomy with a right lateral selective lymph node dissection and parathyroidectomy were performed. Initially, total parathyroidectomy with transplantation of parathyroid tissue was planned, but during surgery, the surgeon could not find the left parathyroid glands, so only two of the right parathyroid glands were removed. The pheochromocytoma were found and special stains were performed to aid in localizing the source of ACTH production. The immunostaining of both pheochromocytoma revealed focal positivity of ACTH staining (Fig. 5). ACTH immunoreactivity was absent in the thyroid carcinoma tissue. The two parathyroid glands were confirmed to have parathyroid hyperplasia. The biopsy revealed no lymph node metastasis from the medullary thyroid carcinoma. After surgery, the hyperglycemia and hypertension were well controlled without medications. Twelve days after the surgery, the ACTH and cortisol levels were 8.5 pg/mL and 0.37 µg/dL, respectively (Table 2). The normal circadian fluctuation was restored with corticosteroid replacement therapy.
Discussion
Multiple endocrine neoplasia 2A is an autosomal dominant syndrome characterized by medullary thyroid carcinoma (MTC) in 90% of affected adults, unilateral or bilateral pheochromocytoma in 50%, and multigland parathyroid tumors in 20~30%. It is the most common form of MEN type 2 syndromes (55% of all cases)[1]. MTC is generally the first manifestation of MEN 2A and develops between 5 and 25 years of age. In older MEN 2A series, in patients with treatment initiated after the identification of a thyroid nodule, MTC progressed and had a 15~20% cancer mortality[5]. Germline activating mutations in the RET proto-oncogene, located on 10q11, were identified as the disease-causing mutation associated with these inherited syndromes[6,7]. This gene encodes a plasma membrane-bound tyrosine kinase enzyme, mainly expressed during development in a subset of neural crest derivatives and in the embryonic kidneys[8]. In 1994, activating mutations of RET were identified and the clinical diagnosis of MTC syndrome carriers, with direct DNA analysis, became available. Subsequently, a consensus was reached at the MEN97 workshop, to perform thyroidectomy in MEN 2 based predominantly on the results of RET mutation testing, rather than calcitonin testing[9]. Currently, early thyroidectomy likely has reduced the mortality from hereditary MTC to less than 5%, including MEN 2A[5].
Cushing's syndrome, caused by ectopic adrenocorticotropic hormone (ACTH) secretion, is most often due to malignant tumors such as small cell carcinoma of bronchus, and cases that are more indolent may be present in patients with underlying neuroendocrine tumors such as bronchial carcinoids endocrine tumors have been responsible for only a small percentage of cases. Wajchenberg et al[4] reported that 2~25% of overt cases of ectopic ACTH secreting tumors were caused by pheochromocytoma, and 2~6% were due to medullary thyroid carcinomas. In Korea, Hong et al[3] was the first to report a case of pheochromocytoma associated with ectopic ACTH syndrome in 1997. However, this report constitutes the first confirmed case of a patient with MEN 2A and Cushing's syndrome with ACTH production from pheochromocytoma in Korea.
In 1988, Mendoca et al[10] first described Cushing's syndrome due to ectopic ACTH secretion with bilateral pheochromocytoma in MEN 2A. Subsequently, the diagnosis and therapy of multiple endocrine neoplasia type 2 has been revolutionized by identification and testing of the RET proto-oncogene mutation. In MEN 2A, early genetic diagnosis, before the development of clinical tumors, is crucial for the cure of this disease. After routine RET analysis, Adriana et al reported a patient with MEN2A and Cushing's syndrome due to an ACTH producing pheochromocytoma[11]. Since then several similar cases have been reported[12~14] however, the number of reported cases is limited. One retrospective series from the French tumor registry found 10 patients with ectopic ACTH production among 1637 patients (0.6%) with diagnosed MTC three out of 10 had MEN 2A, and the pheochromocytoma of two of these patients may have been secreting ectopic ACTH[15].
In the case reported here, the ACTH levels were not remarkably elevated, and the patient did not have typical Cushing features. However, the serum cortisol and plasma ACTH circadian rhythm were disturbed and the serum cortisol was not suppressed after a challenge with high dose dexamethasone. In agreement with the laboratory data and the suspicion of an ectopic ACTH secreting tumor such as a pheochromocytoma or medullary thyroid carcinoma, we excluded Cushing's disease and planned surgery. The immune staining of the tissues obtained at surgery was used to confirm the diagnosis. Immunohistochemistry of the tumor with ACTH confirmed the etiology of the ACTH secretion.
Impaired glucose tolerance has been observed in patients with pheochromocytoma with an incidence of from 25~75%. Decreased insulin secretion is considered the main cause and catecholamines can induce insulin resistance[16]. Such changes in glucose homeostasis improve substantially only after removal of the tumor[17], as observed in this case.
There is a statistically significant association between the presence of the mutation at codon 634 and the presence of hyperparathyroidism[18], as illustrated in this case. The incomplete removal of the parathyroid glands will be monitored with regular follow up of the serum PTH and calcium levels in our patient. This patient did not have a specific family history to suggest MEN 2A in other family members, therefore we inferred a sporadic mutation in this case. However RET proto-oncogene analysis should be performed on other family members to rule out a familial form.




 XML Download
XML Download