

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Pseudohypoparathyroidism (PHP) is a hereditary disorder that resembles hypoparathyroidism, but it limits intact parathyroid hormone (PTH) control on calcium by the resistance against PTH action in target tissues.
In 1942, Albright F and associates[1] described PHP as disease in patients that showed Albright's hereditary osteodystrophy (AHO) such as short stature, round face, short neck, truncal obesity and brachydactyly. These individuals experienced convulsion as well as contracture. Biochemically, hypocalcemia and hyperphosphatemia was observed. Moreover, recent studies reveal some of cases of PHP that show the variability of clinical and biochemical manifestation without AHO findings[2]. There are two different types of PHP distinguished by urinary cyclic AMP (cAMP) response to PTH challenge test and subtypes of these are being researched on the genetic backgrounds. However, there is not enough information about reaction in the PHP associated with other conditions that may cause hypocalcemia.
Rhabdomyolysis is the breakdown of muscle fibers with leakage of potentially toxic cellular contents into the systemic circulation. The etiologies may be diverse and biochemical sequelae include calcium deposition, hyperphosphatemia, impaired calcitriol synthesis, and skeletal resistance to PTH. In this case, we report that rhabdomyolysis caused by infection might have aggravated the hypocalcemia of PHP, resulting in severe hypocalcemia and tetany.
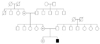
In the past, the genetic mode of PHP was thought to be complex, showing sex linked dominant, autosomal dominant and recessive pattern. But recent studies confirm the GNAS gene presents imprinting phenomenon, explaining the variations in phenotype depending on the maternal or paternal mutation's origin in PHP type I[3]. Three promoters present models of allele-specific methylation and monoallelic transcription. The region NESP55 (neuroendocrine secretory protein 55) is expressed exclusively by maternal allele, while XLas (extra large as like protein) and 1A (1 alternative) are expressed by the paternal allele. Though not yet surveyed in the genetic mutations, our report is the first PHP family by maternal inheritance in Korea.
Case Report
Present illness
A 21-year-old man was admitted to the hospital through the emergency room.
The patient had been admitted 3 weeks ago because of cellulitis on the left ankle at local clinics. He had experienced squeezing pain and contracture in his extremities and felt a fit of tetany which lasted for about one minute without mental loss.
Past history
He had a history of delayed secondary growth and mental retardation but had never been underwent any specific test in details.
Family history
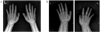
AHO phenotypes appeared through the three generation in the family (Fig. 1). The constellation of findings shown by the patient's mother, sister, and grandmother included short neck, short stature, obesity, round face (Fig. 2A) and shortening on 4th and 5th metacarpal bones in the X-ray (Fig. 3A). Normal mentality was observed.
Physical examination
On the day of admission, the temperature was 36.8℃, the pulse was 102/min, the respirations were 24/min, and the blood pressure was 130/80 mmHg.
On physical examination, the patient appeared short stature and neck. BMI was 38 kg/m2 (height 145 cm, body weight 80 kg) (Fig. 2B). The mental state was drowsy. Carpopedal spasm was observed and Trousseau's and Chvostek's signs were positive. The light reflex in the pupil and deep tendon reflex were normal. There was no sign of cataract and laryngospasm. There were no abnormal findings in the chest and abdomen examination.
Laboratory tests
Laboratory data are shown in Tables 1 and 2. The creatine phosphokinase (CK) and lactic dehydrogenase (LDH) levels were very high, at 8,280 U/L and 2,410 U/L, respectively. Total serum calcium level was 3.7 mg/dL and the serum phosphate level was 7 mg/dL, with PTH level of 17.27 pg/mL. 24-hour urinalysis revealed decrease in urinary excretion of phosphorus and increase in urinary excretion of calcium. 25-(OH) vitamin D level was low, at 8.3 ng/mL.
Biochemistry data of mother was normal except mild increased PTH level of 45.74 pg/mL.
Chest and abdominal radiographs showed no abnormality. Hand X-rays do not show the shortening of metacarpal bones (Fig. 3B). There was marked calcification in basal ganglia on the computerized tomography of the brain (Fig. 4). The rate-corrected QT interval was prolonged on EKG reading.
Decrease of cerebral function was noted in the electroencephalogram and the intelligence test revealed that the score of IQ test was in fifties, which was lower than normal range. Normal karyotype of 46 XY was observed.
Treatment and Course
1st generation cephalosporine was used for cellulitis and fluid was infused for rhabdomyolysis. After calcium gluconate was given intravenously, the contracture was disappeared. Administration of oral calcitrol 1.25 mcg and calcium gluconate 5 g per day was begun. On the 10th day, the CK and LDH levels were normalized, at 475 U/L, 55 U/L. The serum calcium level was 8.8 mg/dL (ionized calcium: 4.38 mg/dL) and the serum phosphate level was 5.1 mg/dL, with an increased parathyroid hormone level of 80.4 pg/mL. An inverse relationship was displayed between serum CK and LDH levels, and serum calcium levels. Although the parathyroid hormone level was 83.9 pg/mL, clinical symptom and biochemical values were improved with the calcium level (8.8 mg/dL) and 25-(OH) vitamin D level (15.8 ng/mL) at discharge and maintained the normal value during three-months follow up.
Discussion
Patients with PHP have elevated PTH levels due to a target tissue resistance. At first, Fuller Albright reported three patients with AHO phenotypes and hypocalcemia in which the administration of parathyroid tissue extracts did not increase in serum calcium or phosphaturia. It was called PHP due to biochemical similarities with hypoparathyroidism, but difference in high PTH levels[1]. In 1969, Chase and cols. revealed there was deficient response of urinary cAMP in patients with PHP and the pathogenesis of this finding should be related to a lack of or defective form of parathyroid hormone sensitive adenyl cyclase in bone and kidney[4].
The PHP presents with low level of serum calcium as well as high level of phosphate. With this fact, we could see that hypocalcemia and hyperphosphatemia cause the specific clinical finding such as tetany and spasm, dental hypoplasia, cataract, calcification of basal ganglia, calcification of soft tissue and well known AHO appearance. About 50% cases of PHP show calcification of basal ganglia[5], like our case.
Hypocalcemia is common during the early phase of rhabdomyolysis, especially when renal failure occurs. Early hypocalcemia is frequently followed by transient hypercalcemia during the diuretic phase of rhabdomyolysis[6,7]. It is suggested that hypocalcemia should result from a combination of factors that include tissue calcium deposition, hyperphosphatemia, impaired calcitriol synthesis, and skeletal resistance to PTH. Calcium binding or accumulation is known to occur. Hyperphosphatemia may be other factors that contribute to hypocalcemia. Decreased synthesis of 1,25-(OH) vitamin D is supposed to occur by the absorption of calcium in necrotic muscle tissue and metabolic acidosis. Llach and associates[8] revealed that decreased synthesis of calcitriol occurred during the hypocalcemic oliguric phase of rhabdomyolysis-induced acute renal failure while increased synthesis of calcitriol occurred during the recovery phase. Massary et al.[9] demonstrated skeletal resistance to the action of PTH during hypocalcemia in renal failure. Hypocalcemia during rhabdomyolysis is usually self-limited and treatment is only considered with caution if hypocalcemic signs or symptoms develop[10].
Three patients with the clinical features of PHP and elevated concentrations of serum CK and LDH was described, to our search: Two cases with PHP type Ia with rhabdomyolysis and one case with PHP type Ib with rhabdomyolysis, all which resulting in tetany[11~13]. So, our case was concluded that symptomatic hypocalcemia might be worsened by the rhabdomyolysis caused by infection of the patient's foot.
The PTH level was normal on admission, but was increased later on phase of disease progression. Hypocalcemia was suspected to increase the PTH value which was not functional. So, hypocalcemia caused rapid progression as well as severe symptoms. After acute phase, PTH level became normalized by supplementary calcium and vitamin D regimens.
There are two different types of PHP based on the mechanism of the renal response to exogenous PTH. Type I can be further divided into 3 subtypes: type Ia, Ib, Ic. Genetic defects associated with different forms of PHP involve the alpha-subunit of the stimulatory G protein (Gsα), a signaling protein essential for the actions of PTH, cAMP transcription and many other hormones.
Type Ia is the most common and first described form by Albright. Patients with this disorder demonstrate AHO appearance and deficient response in urinary cAMP following by administration of exogenous PTH[4,14]. Heterozygous inactivating mutations within Gsα-encoding GNAS exons are found. GNAS gives rise to several different transcripts, including Gsα, XLα (extra-large variant of Gs alpha), and several additional sense and antisense transcripts. The complexity of the GNAS locus is furthermore reflected by a parent-specific methylation pattern. By imprinting phenomenon, it is explained that maternal inheritance of such a mutation, particularly in certain tissues such as proximal renal tubules, leads to PHP-Ia, i.e., AHO plus hormone resistance, while paternal inheritance of the same mutation leads to pseudopseudohypoparathyroidism (PPHP), i.e., AHO only.
There are reports showing deficient action of Gsα activity in RBC, lymphocyte, fibroblast, and platelet and resistance of extraparathyroid cAMP regulating hormone. Farfel et al. tried to ascribe mental retardation in the 50% of type 1a patients to decreased Gsα activity[15].
Patients with PHP type Ib present normal appearance, normal Gsα activity, and isolated resistance to PTH. Although the site and mechanism of genetic defect of type Ib is different with type Ia, hormonal resistance develops only after maternal inheritance of the mutation like type Ia.
Patients with PHP type Ic show AHO phenotypes, normal G-protein activity, resistance to multiple hormones (PTH, TSH, gonadotrophins, and glucagon), and reduced excretion rate of cAMP and phosphate[16].
Patients with PHP type II have normal phenotypes, no resistance to hormones, normal urinary excretion of cAMP after administration of PTH but decrease in phosphaturia.
To the best of our knowledge, fourteen cases of PHP and PPHP have been reported in the Korean literature: AHO phenotypes in seven cases, decreased urinary cAMP after exogenous PTH injection in three cases, no GNAS study. It was classified as six cases with type I, two cases with type II, one case with PPHP. The data of the others was insufficient to be grouped. Particularly, family history of PHP was not observed[17~24]. In our case, AHO phenotypes were inherited through three generations by maternal side. Biochemical abnormalities were found out only to the patient. Identification of mutations in the affected family will be needed.
For differential diagnosis, measurement of 25-(OH) vitamin D levels is important to rule out vitamin D deficiency as a cause of hypocalcemia. In our case, initial 25-(OH) vitamin D level was 8.3 ng/mL, and that was normalized to the value of 15.8 ng/mL. At admission, poor oral intake and low 1,25-(OH) vitamin D3 might have caused transient low 25-(OH) vitamin D level. Moreover, serum phosphorus level is not high in vitamin D deficiency. PTH level is often detectable but inappropriately low and serum calcium levels are only slightly decreased in magnesium deficiency. Chronic renal failure or gastrointestinal loss of calcium can be excluded in our case.




 XML Download
XML Download