

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Chronic obstructive pulmonary disease (COPD) is an increasing chronic respiratory illness characterized by persistent airflow limitation123. The airflow limitation results from pathophysiological changes, especially emphysema, in the airways and lung parenchyme, which are mainly caused by smoking inhalation. However, the mechanism by which smoking provokes emphysema has not been clearly determined, although protease-antiprotease imbalance4, oxidative stress5, and inflammation6 have all been proposed to contribute to the development of emphysema7. Recent studies have reported that apoptosis and autophagy play roles in the pathogenesis of emphysema. An increase in apoptosis of epithelial and endothelial cells in the lungs of COPD patients has been observed8, and studies have shown that several apoptotic models cause emphysema9. Autophagy, an autodigestion in which cytoplasmic materials are delivered to the lysosome10, is one of the processes regulating the apoptosis pathway. Studies have shown autophagy has essential roles in the pathogenesis of neurodegenerative diseases11, and it has been recently highlightened that autophagy has important functions in respiratory diseases12. There have been three forms of distinct autophagy identified: macroautophagy, microautophagy and chaperon-mediated autophagy (CMA)10. In macroautophagy, cytoplasmic components are nonspecifically captured within vesicles to form autophagosomes, which are delivered to the lysosome. CMA transports proteins to the lysosome only via lysosome-associated membrane protein 2a (LAMP2A), a lysosomal transmembrane protein13. While macroautophagy sequestrates cytosolic components in bulk form, CMA targets soluble cytosolic proteins selectively14. The role of macroautophagy in the pathogenesis of COPD has been suggested. Autophagy knockout of microtubule-associated protein 1 light chain 3B (LC3B), the macroautophagic protein, reduced lung apoptosis and airspace enlargement in mice15. However, few studies have evaluated whether CMA could contribute to the regulation of apoptosis. It has been reported that CMA has an opposite activity against macroautophagy14. Thus, we postulated that CMA has a role in the prevention of apoptosis in the pathogenesis of emphysema. In this study, we investigated the impact of autophagy, including both macroautophagy and CMA, on apoptosis in an in vitro model.
Materials and Methods
1. Cells and reagents
Normal human bronchial epithelial cells, BEAS-2B, were cultured in defined keratinocyte-SFM (Gibco by Life Technologies, Grand Island, NY, USA) at 37℃ under 5% CO2. Anti-rabbit LC3B antibody and anti-rabbit caspase-3 antibody were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-rabbit LAMP2A antibody was purchased from Abcam (Cambridge, MA, USA). Anti-poly (ADP-ribose) polymerase-1 (PARP-1) and anti-glyceraldehyde 3-phosphate dehydrogenase were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). All in vitro cell experiments were repeated at least three times.
2. Cigarette smoke extract
Cigarette smoke extract (CSE) was prepared as previously described16. Commercial cigarettes (THIS; 84-mm long, with a diameter of 8 mm; purchased from KT&G, Seoul, Korea) were smoked continuously by a bottle system connected to a vacuum system, and the smoke from 20 cigarettes was bubbled in 60 mL of phosphate-buffered saline (PBS; Gibco). The insoluble particles in the resulting suspension were filtered by a 0.22-µm filter.
3. Western blot analysis
Cellular proteins were extracted using cell lysis buffer (Cell Signaling Technology). The concentration of proteins was evaluated with the Bradford protein assay (BioRad, Hercules, CA, USA) according to the manufacturer's instructions. Equal amounts of protein were resolved by gradient sodium dodecyl sulfate-polyacrylamide gel electrophoresis (Invitrogen, Carlsbad, CA, USA) and transferred to nitrocellulose membranes (GE Healthcare Bio-Sciences, Piscataway, NJ, USA). The membranes were blocked with 5% skim milk, PBS, and 0.1% Tween 20 for 1 hour before overnight incubation at 4℃ with the primary antibodies. The membranes were then washed out three times and incubated with horseradish peroxidase-conjugated secondary antibodies in blocking buffer for 1 hour. After successive washes, the membranes were developed using SuperSignal West Pico Chemiluminescent kit (Thermo Scientific, Waltham, MA, USA).
4. Analysis of cell apoptosis
Apoptosis was determined using an Annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit (BD Biosciences, San Jose, CA, USA). The cells were washed twice with cold PBS and then resuspended in binding buffer at a concentration of 1×106 cells/mL. Five microliters of Annexin V-FITC was added to the suspended cells. After incubation for 15 minutes at room temperature in the dark, the percentage of apoptotic cells was analyzed by flow cytometry.
5. Transfection of small interfering RNAs
Transfection of small interfering RNAs (siRNAs) targeting the LC3B (Cell Signaling) or LAMP2 genes (Santa Cruz Biotechnology) was carried out using Neon Transfection System (Thermo Fisher Scientific) according to the manufacturer's specifications. After 48 hours of transfection, the cells were used in the experiments.
6. Animals
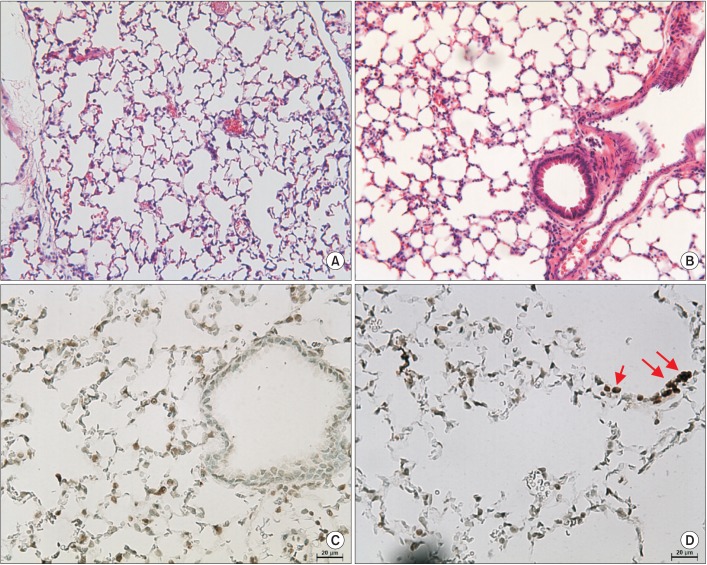
Female 8-week-old C57BL/6 wild-type mice (OrientBio, Seongnam, Korea) were anesthetized and injected intratracheally with 100 µL of CSE (n=3) or buffered saline (control, n=3) once a week for 3 weeks. Recently, we showed the emphysema development in mice by the intratracheal CSE injection16. The mice were sacrificed at 4 weeks, and the lungs were fixed with 4% neutral buffered paraformaldehyde. The fixed lung samples were dehydrated, embedded with paraffin, sectioned, and stained with hematoxylin and eosin. Emphysema was quantified by the measurement of the mean linear intercept (MLI)16. Apoptosis was evaluated with terminal deoxynucleotidyl transferase dUTP nick-end labeling staining. The proportion of the number of positive cells in the four randomly selected high power fileds (×400) was compared between groups by chi-square test.
Results
1. Intratracheal CSE injection led to the development of emphysema and an increase in apoptosis in mice
In the in vivo experiment, intratracheal CSE injection produced emphysema in the mice after 8 weeks (MLI, 25±5 vs. 29±11 µm; p<0.001). The injection of CSE also increased the number of apoptotic cells (p<0.05) (Figure 1).
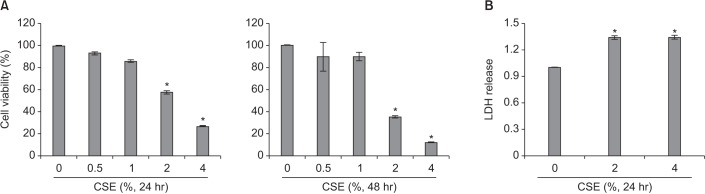
2. CSE decreased epithelial cell survival via an increase in apoptosis
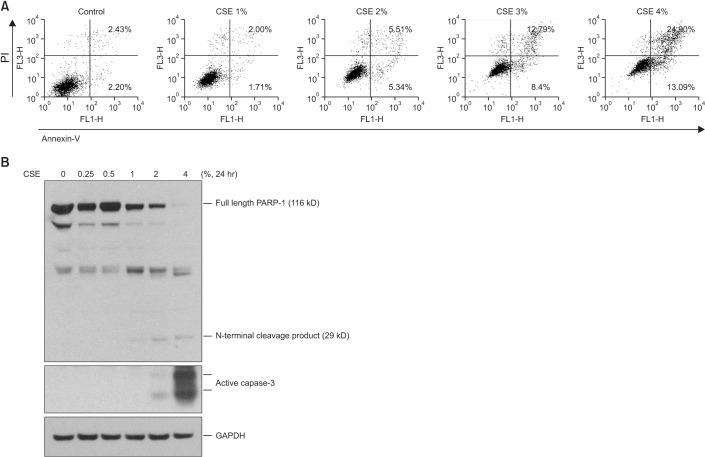
In the 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT) assay, ≥2% CSE significantly decreased the survival of BEAS2-B cells after 24 hours (Figure 2A) and significantly increased the release of lactose dehydrogenase (Figure 2B). The flow cytometry analysis with annexin/propidium iodide staining revealed that CSE increased the apoptosis of BEAS2-B cells in a dose-dependent manner (Figure 3A). Furthermore, ≥2% CSE activated caspase-3 and cleaved PARP-1 (Figure 3B).
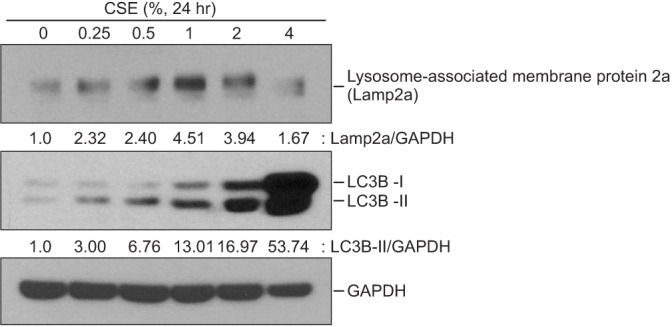
3. CSE increased macroautophagy and CMA in BEAS2-B cells
Both LC3B and LAMP2a expression were increased in cells treated with higher CSE concentrations, indicating that CSE induced macroautophagy and CMA (Figure 4).
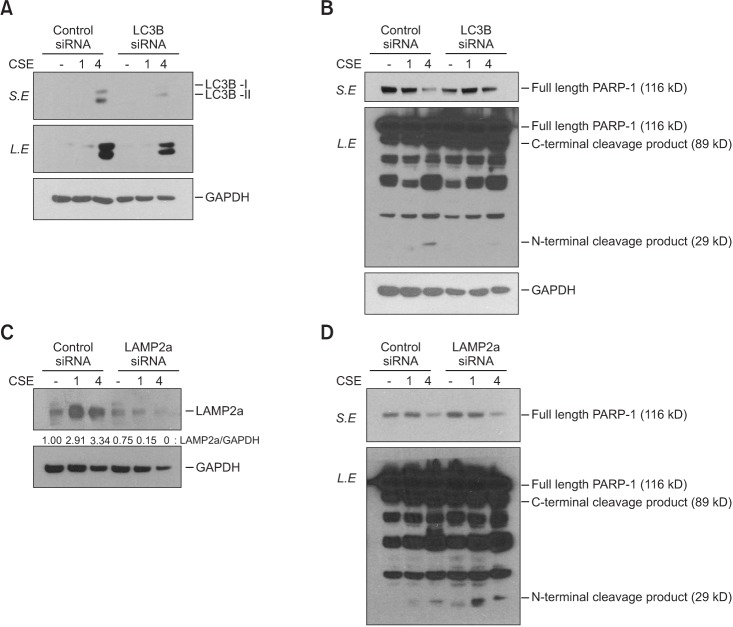
4. Macroautophagy and CMA had different effects on apoptosis in BEAS2-B cells
Both LC3B and LAMP2a expression were knocked down with the respective siRNA treatments (Figure 5A, C). Suppression of LCB3 with siRNA inhibited the cleavage of PARP-1, which suggests that macroautophagy contributes to apoptosis (Figure 5B). By contrast, knockdown of LAMP2a expression led to an increase in PARP-1 cleavage (Figure 5D). These results suggest that macroautophagy and CMA regulate apoptosis in opposite directions.
Discussion
Cigarette smoking is the well-known principal risk factor of COPD. Thus, exposure to cigarette smoke is commonly used to experimentally explore the mechanism and pathogenesis of COPD in vitro or in vivo. Currently, the logical choice of an animal model for COPD is cigarette smoke inhalation, which could result in a pathophysiologic condition similar to that of human patients. However, the lesions induced in the model can be subtle, even upon microscopic evaluation. In addition, it can take as long as 6 months to establish a model17. We developed a new emphysema model of mice, which took only 8 weeks to achieve16. Intratracheal injection of CSE directly into the airway induced parenchymal destruction in the mice. The CSE group showed an increase in apoptosis, which suggested that apoptosis is related to emphysema development (Figure 1). To further explore the contribution of autophagy on apoptosis, we used CSE for an in vitro study.
Although protease-antiprotease imbalance, oxidative stress, and inflammation are regarded as the major contributors to the development of emphysema7, other mechanisms, including apoptosis and macroautophagy, have been studied recently8915. We also demonstrated a relationship between apoptosis and emphysema development in the current study, as described above. Our in vitro study showed the reduction of cell survival when bronchial epithelial cells were treated with CSE (Figure 2), and that increased apoptosis was the major contributor to CSE-induced cell death (Figure 3).
We observed an increase in macroautophagy and CMA in the CSE-treated cells (Figure 4). Previous studies have reported that macroautophagy regulates apoptosis15. However, to date, no study has investigated the role of CMA in the pathogenesis of emphysema. Interestingly, macroautophagy and CMA were found to have opposite effects on cell apoptosis (Figure 5). Macroautophagy increased apoptosis, which has been reported previously. By contrast, CMA suppressed apoptosis according to the results of the siRNA experiment.
The opposite effects of macroautophagy and CMA on apoptosis have been reported previously. If CMA is blocked, macroautophagy upregulation is induced, and vice versa18. For example, Huntington disease cells upregulate CMA activity by increasing LAMP2a de novo synthesis, followed by blockage of macroautophagy19. The upregulation of one autophagic process has been usually regarded as beneficial for cells when the other is compromised20. However, these processes are not completely compensatory. CMA cannot be degraded by particular substrates of macroautophagy and fails to compensate for the macroautophagy-mediated degradation of damaged organelles2122. Interestingly, our results showed that macroautophagy and CMA have reverse effects and lead to contrasting results. However, we should acknowledge that we could not determine the molecular mechanisms of the two autophagic processes.
In conclusion, the intratracheal injection of CSE induces pulmonary emphysema and increases apoptosis in mice. CSE also induces the apoptosis, macroautophagy, and CMA of bronchial epithelial cells. Finally, macroautophagy and CMA regulate apoptosis in opposite directions.
 XML Download
XML Download