

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Tuberculosis (TB) is the major killer disease in many parts of the world. The increase in multi-drug resistant TB, defined as strains resistant to at least two of the first-line TB drugs—isoniazid (INH) and rifampicin and extensively drug resistant TB, has complicated the situation further. Drug resistance in TB is essentially a potential threat to the TB control programs. Of the other types of resistance in TB, INH resistance was found to be higher with 10.3% (new cases) and 27.7% (treated cases) globally1.
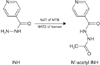
INH is an important first line drug used for the treatment of TB which is highly active against Mycobacterium tuberculosis (MTB). In MTB, the pro-drug INH is transformed to its active form by an enzyme catalase and peroxidase (CP) or KatG. The loss of CP or KatG activities in MTB has long been correlated with resistance to INH23. Multiple genes (katG, inhA, and others) with specific mutations have been associated with INH resistance456. Of these, mutations in katG are responsible for high level resistance and inhA promoter mutations lead to low-level INH resistance. Of all other genes excluding katG in INH resistance, the significance of nat gene coding for N-acetyl transferase (NAT) is worth to note, as it is the sole gene involve in the metabolism of INH in MTB prior to KatG activation of INH to isonicotinic acid7. INH is metabolized by acetylation with the help of NAT in MTB (Figure 1). Also, both KatG and NAT enzymes interact with the pro-drug directly and NAT plays a critical role in mycolic acid synthesis and can be therefore be categorized as efficient anti-TB targets8. Thus, it is of interest to understand contribution of NAT in developing INH resistance in MTB using in silico approaches.
NAT2 in humans is responsible for the inactivation of INH through acetylation similar to NAT of MTB9. The NAT2 of human has long been identified as drug metabolising enzyme. It represents the first examples of pharmacogenetic variation and its study revealed the role of acetyl-CoA as an acetyl donor. Polymorphism in NAT2 gene amongst human has been known to change acetylation activity10. The phenotypic classification of INH metabolisers as fast, slow and intermediate acetylators is based on genetic polymorphism of NAT2 loci11. The wild type NAT2*4 allele is associated with the rapid acetylator phenotype. Polymorphisms in other alleles namely NAT2*5, NAT2*6, and NAT2*7 are associated with slow acetylator status, and polymorphisms such as NAT2*4, NAT2*12, and NAT2*13 are found in intermediate acetylator phenotypes12. Identification of human NAT2 polymorphisms is therefore very significant for predicting the different effective therapeutic doses of INH in fast and slow acetylator1314. The influence of acetylation rate on INH hepatotoxicity is controversial. Hepatotoxicity is the common adverse effect observed in individuals receiving treatment based on INH. Some early studies suggested that fast acetylators were at higher risk for hepatic injury because they generated more acetyl-INH, which could be further metabolized to other toxic intermediaries1516. However, other studies suggest that slow acetylators were also at potential risk1617. In fast acetylators, more than 90% of the drug is excreted as acetyl-INH, making the INH treatment ineffective, whereas in slow acetylators 67% of the drug is excreted as acetyl-INH17. As INH is rapidly acetylated and excreted in fast acetylators, it is proposed that optimal concentration of the drug (INH) is not available in the lungs or affected tissues to mediate its action. As a result of consistently lower serum concentrations of INH and minimal exposure of INH to MTB, gradual buildup of INH resistance occurs. From the host point of view, rapid acetylators may contribute to certain degree of INH resistance based on NAT2 gene polymorphism18. Hence, to understand the contribution of NAT2 from human in developing INH resistance, interactions between the wild type and polymorphic NAT2 gene was carried out in the present study using in silico approaches.
Although originating from different sources such as MTB (pathogen) and human (host) both enzymes (NAT and NAT2) perform similar function and eventually impart INH resistance. Therefore, it is important to investigate the role of NAT of MTB and NAT2 of human in relation to INH resistance. To our knowledge, this is the first study to show the correlation between NAT of MTB and NAT2 of human in developing INH resistance.
Materials and Methods
1. Protein (homology modeling of mutant NATs)
In the present study, the target protein sequences of NAT (Rv3566c) of MTB and NAT2 of human (P11245) were obtained from the Tuberculist database (http://genolist.pasteur.fr/TubercuList/) and Uniprot database (http://www.uniprot.org/), respectively. The obtained protein sequences of MTB and human were submitted to protein alignment program (BLASTp)19 and searched against protein database (PDB; http://www.rcsb.org/pdb/home/home.do). The WT-NAT and NAT2 protein structures of MTB and human (4BGF and 2PFR) were considered as the templates, respectively2021.
2. Model building and evaluation
Residues at position 207 of the NAT protein of MTB and 268 of NAT2 of human were substituted to generate mutant (MT) proteins. In other words, the MT of NAT of MTB (G207R) was generated using crystal structure (4BGF) of NAT protein of MTB obtained from PDB20. In case of human the MT of NAT2 (K268R) was generated using crystal structure 2PFR, WT-NAT2 protein of human21. In templates 4BGF and 2PFR, A chain was used and the heteroatom such as water was removed, and command line options were provided for sequence alignment between WT and MTs, and then series of commands were provided for model building using software MODELLER9v1422.
3. Ligands
The ligands (acetyl CoA, acetyl INH, and INH) used in this study were obtained from Chemspider database (http://www.chemspider.com). Chemsketch software25 was used to obtain the structure of the ligand in Mol format and the ligand was saved as Mol2 file using software Discovery Studio26.
4. Discovery Studio, docking protocol, and BIOVIA-2016
The Discovery Studio software was used for visualization purpose of modelled proteins, and docking data26.
Docking was carried out with the help of software-GOLD27. The GOLD protocol is based on the principle of genetic algorithm wherein the receptor is held rigid while the ligands are allowed to flex during the refinement process. The input files for both the protein and the ligands were generated. Hydrogen atoms were added to the models and ligands using auto edit option in GOLD before docking followed by energy minimization. The cavity atom file containing the atom number of binding residues was prepared for ligands such as acetyl-INH and INH (Phe38, Tyr69, Cys 70, Val95, Thr109, Phe130, Gly131, and Phe204). The binding residues were selected based on comparison between the binding regions of 4BGF19 and the crystal structure of 3LTW in complex with hydralazine compound of 2.1 Angstrom (Å) resolution28. In case of acetyl CoA, the binding residues such as Cys70, Val 95, Trp97, Leu98, Thr109, His110, Phe130, Gly 131, Gly132, Glu152, Val169, Arg170, Phe204, Met209, Ala210, Arg218, Asn220, Met 222, His229, Gly232, and Thr234 were chosen based on 2VFC29 of 2.7 Å resolution in complex with CoA on comparison with the binding regions of 4BGF.
In case of human, the cavity atom file for acetyl CoA containing the atom number of binding residues was prepared, the residues (Phe37, Cys68, Phe93, Ile95, Pro97, Val98, Ser102, Thr103, Gly104, His107, Leu109, Gly124, Tyr208, Thr214, Ser216, Phe217, Ser287, and Leu288) were directly selected from the binding regions of CoA from 2PFR, because the template was already in complex with the ligand-CoA. Secondly, the binding residues (Phe-37, Trp-67, Cys-68, Ileu-106, His-107, and Phe-217) for acetyl-INH and INH (Phe-38, Tyr-69, Cys -70, Tyr-71, Val-95, Thr-109, Phe-130, and Phe-204) were obtained from crystal structure of 1W6F30 in complex with INH of 2.1 Å on comparison with the binding regions of 2PFR.
Dockings were performed under “standard default settings” mode-number of islands was 5, population size was 100, number of operations was 100,000, niche size was 2, and selection pressure was 1.1. Ten docking poses were obtained for each ligand. Poses with highest GOLD score were used for further analysis. The docked poses of the ligands were visualized using Discovery Studio. The scoring function of GOLD provides a way to rank the ligands relative to one another. Ideally, the score should correspond directly to the binding affinity of the ligand for the protein, so that the best scoring ligand poses are the best binders.
This BIOVIA-2016 software was used to determine the interactions between the ligands and proteins31.
Results
1. Template selection and homology modeling of MTs of NATs
Since G207R and K268R are the two substitutions frequently associated with polymorphism in NAT of MTB and NAT2 of human, respectively. MT models of NAT and NAT2 were built based on substitutions at these codon 207 and 268 in the WT-NAT (Rv3566c) and NAT2 protein sequence (P11245) of MTB and human, using MODELLER 9v14 (Figures 2, 3) respectively. Using BLASTp search against PDB, 4BGF and 2PFR were identified as the templates as it displayed maximum identity with the NAT proteins (refer Supplementary Figures S1, S2 for sequence alignment). The generated models were validated by structural superimposition (Figures 4, 5). The root mean square deviation (RMSD) between the WT and MTs was 0.2 Å in case of MTB-NAT, and 0.1 Å in case of human NAT2 indicating the reliability of the model. This was also supported by Ramachandran plot analysis22 (refer Supplementary Figures S3, S4 for plots).
2. Docking between ligands and NATs of MTB and human
The generated models were used for docking studies, and the docked complexes (Figures 6, 7) were visualized using Discovery Studio. The process of docking was validated using the experimental structure complexed with ligands such as CoA, acetyl-INH and INH with the WT docked by different ligands. In case of MTB, the crystal structure complex with CoA (2VFC) was superimposed with WT docked by acetyl CoA. Secondly, crystal structure complex with hydralazine (3LTW) was superposed with WT docked with acetyl-INH and the RMSD values obtained were 0.5 Å and 0.1 Å for CoA and acetyl-INH, respectively (Figure 8A, B). In case of human, the crystal structure complex with CoA (2PFR) was superimposed with WT docked by acetyl CoA. Interestingly, no deviation was observed; hence, the RMSD value obtained was 0.0 Å. The crystal structure (1W6F) complexed with INH was superimposed by WT protein docked with INH, this showed more extent of deviation of about 0.7 Å of RMSD (Figure 9A, B).
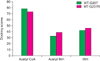
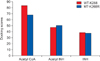
Docking of ligands with NATs resulted in ten poses. Of the 10 poses, the best ligand pose was selected based on top GOLD score. In case of MTB-NAT, the highest score of −93.59 kcal/mol was obtained for between the ligand acetyl CoA and the WT compared to the MT; followed by acetyl-INH and INH. Although, similar pattern was obtained in case of human NAT2 with high score for acetyl CoA when compared to that of the MT, while not much difference in scores was observed with respect to the ligand-INH between WT and MT (Figures 10, 11).
3. Interactions at the ligand binding site in MTB-NATs
Of all type of interactions, hydrogen (H) bond interactions are very significant as it affect the stability and integrity of ligand and protein complex. In case of WT-MTB, H bonds (carbon H) were found between the ligand (acetyl CoA) and Glu152, Met168 residues and conventional H bond was formed between the ligand and residue-His229 of WT-NAT. van der Waals interactions were found more in number compared to other types. Alkyl and pi-alkyl were found at the acetyl CoA binding regions in the WT and as well in MT. Interestingly, all types of interactions including the unfavorable one with Thr234 were found between WT-MTB-NAT and acetyl CoA. Of which, the attractive charge and salt bridge were observed between phosphate groups of acetyl CoA and residues such as Arg170 and Lys236. In case of the MT complex with acetyl CoA, the same interactions (Arg170 and Lys236) were observed in addition to the participation of Lys98 with phosphate groups (Figure 12A).
In case of the ligand-INH complexed with WT, two conventional H bonds between nitrogen (N) atom of INH and one with His151, another with His229; a carbon H bond between His151 and oxygen (O) atom of INH were found. Of note, pisigma, amide-pi stacked and pi-alkyl bonds were formed between pyridine ring of INH and Glu152, Arg230 and Ala231 residues, respectively. In MT, a carbon H bond was formed between the residue His229 and N atom of INH, pi-alkyl bond between Arg218 and pyridine ring of INH. Further, in contrast to WT with pi-sigma bond, a pi-anion bond was formed between Glu152 and pyridine ring of INH. Interestingly, an unfavorable interaction between N of INH and Gln133 (Figure 12B) was observed, which lacks in WT.
In case of the acetyl-INH complexed with WT, many van der Waals interactions were found followed by two conventional H bonds that were contributed from Gly131 to O and N atoms of the ligand. An unfavorable interaction from Lys203 and a pi-cation from Phe130 were also found in WT. On the other hand in case of MT, followed by van der Waals interactions three conventional H bonds were imparted from Thr109 and Phe130, and a carbon H from Gly129 (Figure 12C).
4. Interactions at the ligand binding site in human-NATs
In case of human-NAT2, the interactions were more in case of WT complex with acetyl CoA compared to MT. Following van der Waals interactions, more number of H bond interactions such as seven conventional H bonds and two carbon H bonds were formed. Pi-pi stacked (2) and alkyl and pi-alkyl bonding (5) were also formed. In contrast, in case of MT only a single conventional H bond and two carbon H bonds were formed, followed by a couple of alkyl bonds (Figure 13A).
The WT-NAT2 interacts with INH in a different manner with no conventional H bonding and a single carbon H bond, followed by the common van der Waals interactions. Importantly, pi-sulphur interaction was found between pyridine ring of INH and Cys68. Similarly, pi-pi stacked was formed between pyridine ring of INH and Phe93, and pi-pi T-shaped was formed between pyridine ring of INH and Phe217 of WT. Interestingly, in case of MT similar type of carbon H bond interaction as that of WT was found between Val106 and pyridine ring of INH. Moreover, two conventional H bonds were found between N atom of pyridine ring in INH and one with Cys68; and another with His107 of MT, respectively. Further, pi-pi stacked similar to that of WT and pi-alkyl interactions between Cys68 and pyridine ring of INH were also found (Figure 13B).
The WT complex with acetyl-INH showed interesting types of interactions such as pi-sulfur, pi-pi T-shaped, pi-pi stacked and pi-alkyl between aromatic nucleus and Cys68 residue, Phe217, Phe93, and Val106, respectively. Importantly, two picationic bonds were formed between two N atoms of pyridine ring in INH and one with Phe37; another with Phe97 of WT, respectively. Single conventional H bond and carbon H bond were present, as shown in figure. On the other hand, in case of MT, 5 conventional H bonds were formed. Similar to WT, pi-pi stacked and pi-pi T-shaped, pi-alkyl bonds were formed between MT residues and pyridine ring of INH, respectively (Figure 13C).
Discussion
The enzyme NAT is responsible for the transformation of the active form of INH (isonicotinic acid hydrazide) to its inactive counterpart (acetyl-INH) in the presence of the (cofactor) acetyl CoA by transferring acetyl moiety from acetyl CoA to INH, in MTB. Similarly, in case of human, NAT2 is also responsible for the inactivation of INH through the process of acetylation. Despite different origins, the evolutionary conserved NAT enzymes perform similar function that is the conversion of INH to acetyl-INH. In case of MTB, MT-NAT mediates INH resistance by the formation acetyl-INH through competing KatG, which is unable to perform the normal function of activation of INH. Thus, if NAT excels KatG it would mediate INH resistance; on the other hand if KatG excels NAT, it would result in INH susceptibility8.
In case of human-NAT2, NAT2*4 polymorphism as evident in fast acetylator is responsible for INH resistance. In addition to NAT2*4 , NAT2*12 and NAT2*13 alleles of intermediate phenotypes are also associated with rapid acetylator. In NAT2 gene, polymorphisms at nucleotide positions 282, 481, and 803 are responsible for rapid and intermediate acetylating phenotype. Of these, polymorphism (TAC-TAT; C to T) at nucleotide positions 282 and 481 result in silent mutation with Tyr94Tyr and Tyr160Tyr conversion. Interestingly, polymorphism (AAA-AGA; A to G) at position 803 of NAT2 gene with substitution of Lys to Arg leads to conformational changes in the NAT2 protein at codon 26818. The frequency of rapid acetylator genotypes in the Japanese is much higher than in the Caucasians population14. Rapid acetylators may not respond to INH treatment efficiently and could be a potential risk factor for the development of INH resistance in humans. Since INH is rapidly acetylated in fast acetylators, which eventually leads to INH resistance could be due to lower serum concentrations of INH and minimal exposure of INH to MTB in affected tissues18. The recommended higher dose of INH of 7.5 mg/Kg instead of standard dose (5 mg/kg) can overcome INH resistance without causing adverse side effects to the patients in case of rapid acetylators14.
Taking into account of the biological significance of these enzymes, in this study, the WT and MT-NATs of MTB and human were modeled and docked with substrates and product (acetyl CoA, acetyl-INH, and INH) with insight to find differences in the binding ability between WT and MT. Based on the results of docking in MTB-NAT, it can be suggested that the binding affinity of MT-G207R with acetyl CoA was found to be less and more with acetyl-INH and INH compared to the WT. In other words, the high docking score with acetyl CoA could be due to presence of favorable interactions, in WT. On the contrary, the MT showed less affinity towards acetyl CoA than the WT, perhaps due to the presence of unfavorable donor-donor interactions and non-formation of unfavorable positive-positive and acceptor-acceptor interactions which were evident in WT. Since, MT shows more affinity towards acetyl-INH; on the basis of which, it can be speculated that the process of acetylation could occur at a faster rate, leading to high rate of formation of acetyl-INH through inactivating INH and this may result in INH resistance eventually.
In case of human NAT2 also, the pattern of score was similar to MTB-NAT with respect to acetyl CoA (decrease in the binding affinity in MT-K268R) and increase in acetyl-INH, but decrease in INH score inspite of presence of more number of H bonds. The reason for the high score in the WT with acetyl CoA was owing to the presence of more number (7) of conventional H bonds and also alkyl bonds (5) compared to less numbers in MT. Increase in case of acetyl-INH due to more number of (5 and 2) of conventional H bonds in MT compared to WT. Thus, in MT, acetylation occurs at a faster rate and formation of acetyl-INH speeds up serving as possible risk factor for developing INH resistance in human in rapid acetylators. However, the effect of docking in both MTB and human could be better explained after performing molecular dynamics for understanding their function precisely. Nevertheless, the information provided over here can be useful to understand the impact of such substitution and consequent changes in binding ability. More importantly, it can be suggested that the phenomenon of emergence of INH resistance is not unidirectional from MTB alone rather it's a bidirectional phenomenon, derived from both pathogen (MTB) to a major level and host (human) to a lesser extent. In order to better understand the scenario of INH resistance with respect to contribution from host and pathogen further molecular studies are warranted pertaining to sequencing of nat gene in clinical isolates of MTB, isolated from those individuals in whom NAT2 genotyping was performed to identify rapid acetylators. Further, as the MT-NAT and NAT2 enzyme from MTB and human commonly have shown increased binding affinity to its product, acetyl-INH, respectively. An approach based on clinical prediction of rapid-acetylating N-acetyltransferase could be to determine the accelerated binding affinity towards the product, contributing to INH resistance, provided these MT enzymes are known to be rapid acetylators. Such diagnostic finding is worthwhile to be tested in future studies with various other clinical mutants of NAT.







 XML Download
XML Download