

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Streptomycin (SM) is the first anti-tuberculous drug that combated efficiently to tuberculosis (TB). Owing to the high rates of resistance due to mono-therapy and toxic side effects, the usage of SM as the first option for TB has been gradually declined. To date, it is recommended by World Health Organization (WHO) in standard regimens for the retreatment and multidrug-resistant tuberculosis (MDR-TB) cases1.
SM binding to bacterial ribosomal protein S12 and 16S rRNA encoded by rpsL and rrs, respectively, which locate in the decoding center of 30S ribosomal subunit interrupts the protein translation fidelity, thereby inhibiting protein synthesis and resulting in bacterial death2. Consequently, rpsL and rrs mutations confer the high or intermediate SM resistant levels in Mycobacterium tuberculosis34. The rpsL 128A>G (Lys43Arg) and single nucleotide polymorphisms (SNPs) in areas of the 530 and 912 loops of rrs are the most frequently found mutations in SM resistant isolates56. Furthermore, the low-level SM resistance (SMR) in M. tuberculosis is caused by mutations in a 7-methylguanosine (m7G) methyltransferase (gidB) gene which functions for methylation of G527 in 530-loop of 16S rRNA5. The frequencies and types of SMR associated mutations in the above-mentioned genes are dependent on the geographic areas and M. tuberculosis lineages678. In addition, some gidB SNPs are responsible as M. tuberculosis lineage signatures rather than SM resistant markers68. Approximately 30% of SM resistant strains worldwide carried no mutation in those three genes and therefore, the contribution of alternative genes conferring intrinsic mechanism of SMR such as aminoglycosides efflux pump Tap (Rv1258c) and its transcriptional regulator, whiB7 (Rv3197A), have been suggested 491011.
Thailand is one of the 22 highest TB burden countries and the incidence of MDR-TB in new and retreatment cases were 2% and 19%, respectively in 201412. However, to date, the information on M. tuberculosis mutations associated to SMR in Thailand is limited to only one study8. Therefore, we further expanded the investigation on the mutations of rpsL, rrs, gidB, and whiB7 in M. tuberculosis isolates collected from the different time and region of Thailand, and associated with their SMR and lineages.
Materials and Methods
1. M. tuberculosis isolates, drug susceptibility testing, and spoligotyping
A total of 287 M. tuberculosis isolates collected during 2007 to 2011 were obtained from Bamrasnaradura Infectious Diseases Institute, Nonthaburi, Thailand. Their drug susceptibility testing (DST) was tested for four first-line anti-TB drugs using absolute concentration method on Löwenstein-Jensen medium with following concentrations, 0.2 and 1 µg/mL for isoniazid (INH), 40 µg/mL, 4 µg/mL, and 2 µg/mL for rifampicin, SM, and ethambutol, respectively13. Among those, 46 isolates were found SM resistant. These and other 55 SM susceptible isolates in the same collection were further analyzed for mutational profiles. Their lineage and strain type (clade) were identified by standard spoligotyping protocol and TB lineage online tool1415. Details of anti-TB drug susceptibility phenotypes and lineage differentiation of 101 isolates were shown in Table 1. The pan anti-TB drug–susceptible M. tuberculosis H37Rv (ATCC 27294) strain was used as a reference in all assays.
2. Amplification and sequencing of rpsL, rrs, gidB, and whiB7
Genomic DNA was extracted from 101 M. tuberculosis clinical isolates by the cetyltrimethyl ammonium bromide method as previously described16. Oligonucleotide primers used for polymerase chain reaction (PCR) amplification and sequencing of rpsL, rrs, gidB, and whiB7 were based on previous studies or newly designed in the present study (Table 2)1718192021. Amplifications were carried out using Pfu DNA polymerase (Bioneer Corp., Daegeon, Korea). Sequencing was done in both forward and reverse directions using BigDye Terminator Cycle Sequencing Kit version 3.1 (Applied Biosystems, Foster City, CA, USA) in an ABI 3730XL DNA analyzer (Life Technologies, Applied Biosystems). DNA sequencing data were analyzed and assembled using Sequence Scanner v2.0 (Applied Biosystems) and DNAMAN (version 4.15, Lynnon BioSoft, Quebec, Canada) software. Mutations were identified by comparing to the wild type (WT) M. tuberculosis H37Rv reference sequence (accession No. AL123456.3) (Table 3)22.
3. Mismatched amplification mutation assay PCR for detection of mutation at rrs nucleotide position 16
Due to our rrs sequencing data could not reveal the first 39 nucleotides of the coding sequence, mismatched amplification mutation assay PCR (MAMA-PCR) was developed to detect the recently identified 16T>C (16>c for RNA) mutation of rrs sequence (Table 2). MAMA-PCR primers were designed in this study to differentially detect T to C nucleotide alteration at nucleotide position 16 of rrs (previously annotated as nucleotide position 15)2324. As shown in Table 2, the mutant allele-specific forward primer (rrs-16 mt) contained mutated nucleotide ‘C’ instead of WT ‘T’ at the 3'-end. The rrs-RM primer was used as a shared reverse primer. The amplification of nucleotide position 16 mutant allele present exclusively in reactions containing rrs-16 mt primer but not rrs-16 wt and vice versa for the WT allele. The genomic DNA of H37Rv was used as a control for WT allele amplification. Mutant allele control, 16T>C rrs was constructed by overlap extension PCR (data not shown).
4. Verification of Beijing isolates by RD105 deletion-targeted multiplex PCR
In order to verify the spoligotyping results, RD105 deletion-targeted multiplex PCR (DTM-PCR) was performed for all Beijing (East Asian) isolates (n=57) and representatives of non-Beijing isolates (n=18) as previously described21. The non-Beijing isolates carrying intact RD105 yielded approximately 1,495 bp amplicons while the Beijing isolates (including ancestral, modern, and W family of Beijing lineage) containing a 3,467 bp deletion of RD105 region yielded only 785-bp DTM-PCR product.
5. Statistical analysis
The association of identified polymorphisms to SMR and M. tuberculosis lineages was assessed with GraphPad Prism 6 software (Graphpad Software Inc., La Jolla, CA, USA). The p-value (<0.05) was calculated by Fisher exact test. Odds ratio (OR), 95% confidence intervals (CI), sensitivity, and specificity were also calculated.
Results
1. SMR associated genes' mutations and their relationship to SMR
The entire length of rpsL and gidB, and 1,528 bp of rrs were amplified and sequenced from genomic DNA of 101 M. tuberculosis isolates. As shown in Table 3, the majority of SM resistant isolates, 32/46 (69.6%), carried rpsL 128A>G transition (Lys43Arg). Among them, mixed rpsL WT and 128A>G sequences were found in a SM mono-resistant EAI2-Nonthaburi isolate. Other two rpsL missense mutations, i.e., 263A>G (Lys88Arg) and 263A>C (Lys88Thr), were identified in two SM resistant MDR isolates. Lys43Arg was the most predominant rpsL mutation (94.1%) among SMR isolates in this study. However, we found 2/55 SM susceptible isolates carrying Lys43Arg.
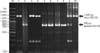
Surprisingly, none of rrs mutation related to SMR was present in M. tuberculosis isolates from our collection. Only a single rrs SNP, 1025u>c, was found in a SM susceptible isolate (Table 3). Further examination of the previously found 16T>C SMR related rrs mutation by MAMA-PCR newly developed in this study indicated that none of our isolates carried that respective mutation (Figure 1).
Six different types of gidB SNPs were identified. Among them, two SNPs were exclusively found in two Euro-American isolates with SMR. The 134G>A (Trp45Ter) was carried by a SM mono-resistant isolate lacking rpsL mutation, whereas, the 206G>A (Gly69Asp), a novel gidB mutation identified in this study, was found in a SM resistant MDR isolate carrying Lys88Thr rpsL (Table 3). Other four gidB SNPs as presented in Table 3 were unlikely to associate with SMR because they were identified in both SM resistant and susceptible isolates.
The nucleotide sequence of the entire whiB7 ORF and its promoter region were further analyzed in 12 SM resistant isolates (26%) lacking of SMR-associated rpsL and rrs mutations. Two SNPs in the ORF region, i.e. 242G>A (Arg81His) and 246_247insTT (Arg83fs), were found in a SM/INH resistant East Asian isolate (Tables 1, 3). A frameshift mutation (188delG) was present in three Indo-Oceanic (EAI2-Nonthaburi) isolates (Tables 1, 3). BLAST analysis revealed that their deduced amino acid sequences were identical to ‘disrupted’ efflux pump activator from EAI2-Manila lineage M. tuberculosis25. Thus, 188delG mutation was unlikely to relate to SMR.
Among three SMR-associated genes, rpsL mutations were highly associated to SMR (73.9%) (p<0.001). The combined detection of three rpsL and two gidB mutations provided 76.1% sensitivity and 96.4% specificity for identification of SMR in Thai M. tuberculosis isolates. Similar to another Thai study8, SMR was strongly associated to East Asian (Beijing lineage) (32/46 isolates, 69.6%) (OR, 2.7; 95% CI, 1.2–6.2; p=0.017) (Table 3). Moreover, rpsL mutations were predominant among SM resistant Beijing isolates (93.8%) (OR, 37.5; 95% CI, 5.9–236.6; p<0.001) while their presence was significantly lower among non-Beijing lineages (28.6%). However, in this study, no significant correlation between SMR or SMR-associated mutations and MDR phenotypes was found (p=0.859 and p=0.112, respectively). Furthermore, there was no relationship between the MDR isolates and lineage of M. tuberculosis in our collection (data not shown).
2. Polymorphisms of gidB and whiB7 as phylogenetic signatures for M. tuberculosis lineage
Regardless of SMR, 615A>G gidB was found in all East Asian and 32/34 Indo-Oceanic isolates in our study (Table 3). Whereas, the 276A>C (Glu92Asp) gidB, a known marker for the Beijing isolates68, presented in 55/57 spoligotyped-Beijing (East Asian) and 2/34 EAI5 (Indo-Oceanic) isolates (Tables 1, 3). The discrepancies were further clarified by DTM-PCR, and it was found that the 55 Beijing and 2 EAI5 isolates with Glu92Asp carried RD105 deletion specific for Beijing lineage (Figure 2). In contrast, one of two spoligotyped-Beijing isolates lacking Glu92Asp carried the intact RD105 region while another carried mixed intact and deleted RD105 genotype (Figure 2).
In addition to 615A>G gidB, the 26/29 Indo-Oceanic (EAI) isolates in this study also carried 330G>T gidB silent mutation (Tables 1, 3). Interestingly, the two spoligotyped-Beijing isolates lacking Glu92Asp SNP but with intact RD105 region mentioned above, also carried the EAI marker, 330G>T gidB. Five of the Indo-Oceanic isolates, all exclusively belonging to EAI6-BGD1 sub-lineage, carried triple silent mutations (423G>A, 330G>T, and 615A>G) (Tables 1, 3), demonstrating the potentiality of 423G>A gidB SNP as an additional novel marker for EAI sub-lineage classification. Moreover, a whiB7 frameshift mutation (188delG), unlinking to SMR, appeared to be present exclusively in the three Indo-Oceanic sub-lineage EAI2-Nonthaburi isolates25.
Discussion
The amino acid lysine at codons 43 and 88 are the key SM binding sites on the ribosomal protein S12 coded by rpsL26. Therefore, any substitution to lysine such as Lys88Arg (or Lys43Arg or Lys88Thr) would ultimately result in SMR of bacteria including M. tuberculosis as a consequence of the reduction in SM binding activities. The majority of SMR in M. tuberculosis isolates in this study was contributed by the rpsL mutations (73.9%). Although, this mutation frequency was higher than that of the average worldwide frequency (52%–59%)4 and a single previous report from Thailand (63.6%)8. However, it was still lower than that from China, Vietnam, and Singapore72728. Similar to those reported from South East Asian countries, the most predominant rpsL mutation associated to SMR in this study was Lys43Arg (69.6%)82728. As the mutation in either codon 43 or 88 of rpsL had a strong connection to high-level SMR3, the majority of SM resistant M. tuberculosis isolates in our collection would possess the same level of SMR. Furthermore, the mutation in these two codons of rpsL markedly predominated among Beijing isolates of our study (93.8%)2728.
It was found that 2/55 SM susceptible Beijing isolates carried Lys43Arg mutation. Previously, the genotype-phenotype discrepancy of SMR was verified by performing DST on M. tuberculosis isolates collected from the subsequent follow-up sputum samples of the same patients and it was then proved that the newly collected isolates had become SMR29. These data suggested the significance of re-performing DST on the isolates from the follow-up sample of a patient who was originally infected with SM susceptible M. tuberculosis carrying Lys43Arg rpsL that might exhibit the SM hetero-resistant phenotype. However, the follow-up samples were not available in this study.
Surprisingly, none of SMR-related rrs mutations was present in M. tuberculosis isolates from our collection although the only previous study of M. tuberculosis Thai isolates collected from the different time and region revealed that 8.2% carried SMR-associated mutations in 530 and 900 regions of rrs8. The prevalence of rrs mutations responsible for SMR in this bacterium might be geographically dependent having with 0% for India and Sierra Leone3031, and less than 10% in East and South East Asian countries78272832. While, European and Latin American countries reported the higher prevalence (13%–28%)6333435. Although our sample size was smaller than that of the previous study from Thailand, the proportions of isolates with SMR (16% compared to 10%) and rpsL mutations-associated SMR (73.9% compared to 63.6%) was apparently higher8. Hence, it was possible that the absence of SMR-related rrs mutations was an outcome of the upsurge in SMR-related rpsL mutations. Moreover, our data implied the possibility that the genetic background of SM resistant M. tuberculosis isolates collected from the different period and regions of Thailand might be substantially diverged.
Only two of six gidB mutations, Trp45Ter and Gly69Asp were likely to be responsible for SMR and exclusively found in Euro-American isolates. This finding coincided with the suggestion of the previous study that the isolates harbored only SMR-associated gidB mutations without those of rpsL and rrs were non-Beijing8. However, currently, the contribution of a novel gidB mutation found in our study, Gly69Asp, to SMR was unclear as this isolate also carried Lys88Thr rpsL.
For 12 SM resistant isolates missing of rpsL and rrs SMR-related mutations, three mutations, i.e., Arg81His, Arg83fs, and Gly64fs were found in the ORF of efflux pump activator, whiB7. Thus, they would contribute to the SM hyper-susceptibility rather than the SMR phenotype9. As a consequence, after excluding the gidB Trp45Ter mutant, the genetic constituents responsible for the SMR in 11 SM resistant isolates (24%) in the present study remained unknown. As a frameshift mutation, Gly64fs (188delG) was present in all three whiB7-sequenced Indo-Oceanic (EAI2-Nonthaburi) isolates and the EAI2-Manila sublineage from the Genbank database25, the availability of this mutation as sub-lineage marker required further elucidation.
We found that SMR and its associated rpsL mutations were strongly linked to the Beijing lineage (p=0.017 and p<0.001, respectively), whereas there was no relationship between MDR isolates and the SMR or Beijing isolates. Therefore, the WHO recommendation for incorporation of SM as a standard regimen for retreatment and MDR-TB cases in this region would remain fully effective as long as the SMR-associated mutations and Beijing lineage differentiation were performed as a prerequisite.
Among four SMR-unrelated gidB SNPs identified, the Glu92Asp was the well characterized Beijing lineage-specific marker568. Moreover, the presence or absence of Glu92Asp and the carrying of RD105 deletion or intact types were better correlated than the spoligotying. It had been suggested that the phylogenetic accuracy of direct repeats (DRs) locus genotyping methods such as spoligotyping and MIRU-VNTR exhibited lesser discriminatory power for lineage classification compared to the gold standard DNA sequencing36. Accordingly, SNPs which bear almost no homoplasy have become more dependable genetic markers for phylogenetic differentiation in M. tuberculosis. Therefore, either the RD105 deletion in the same fashion as Beijing genotype in the EAI strains or retaining of intact RD105 in Beijing strains strongly suggested the presence of the convergent evolution between Beijing and EAI M. tuberculosis Thai isolates.
Furthermore, the 26/29 Indo-Oceanic isolates in our collection were EAI isolates and carried 330G>T gidB silent mutation, highlighting its specificity as a phylogenetic marker for EAI strains831. Interestingly, all five EAI6-BGD1 SpolDB4 family isolates belonging to Indo-Oceanic lineage carried triple silent mutations (330G>T, 423G>A, and 615A>G), demonstrating the potential of 423G>A gidB as a novel marker for EAI sub-lineage classification. Although the presence of this mutation was previously shown in 6/31 EAI M. tuberculosis Thai isolates, their association to the SpolDB4 family sub-lineage was not made8. However, its applicability as the EAI sublineage identification required further studies on the larger population of M. tuberculosis isolates.
In summary, we investigated the sequences of rpsL, rrs, and gidB in 101 M. tuberculosis Thai isolates to identify the mutations conferring SMR. The three rpsL SMR-associated mutations were found in 73.9% of SM resistant isolates suggesting that they possessed a high-level of SMR. Further analysis of the whiB7 sequence in the SM resistant isolates lacking rpsL and rrs mutation revealed only the SNPs within whiB7 ORF which could not confer intrinsic SMR. The present study confirmed the usefulness of four gidB mutations as a lineage phylogenetic marker for East Asian (Beijing), Indo-Oceanic and Euro-American. Moreover, 423G>A was identified in this study as a new putative surrogate makers for EAI subclade (EAI6-BGD1).




 XML Download
XML Download