

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Cigarette smoking is the major risk factor of chronic obstructive pulmonary disease (COPD)1,2. As a major component of cigarette smoke, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, dioxin) and TCDD-like chemicals cause a wide range of pathologies3. In addition to airway epithelial cells, many inflammatory cells play a major patho-physiological role in asthma and COPD4,5,6,7,8. These inflammatory cells contribute to the activation of inflammatory cytokines including transforming growth factor (TGF) α, TGF-β, interleukin (IL) 1β, IL-6, IL-8, and interferon γ9,10,11. Aryl hydrocarbon receptor (AhR) is a ligand-dependent transcription factor that binds to a wide variety of synthetic and naturally occurring compounds and is involved in regulating inflammatory responsiveness during acute and chronic respiratory diseases12,13. The prototype of AhR ligand is the TCDD, which has a broad range of toxic effects. Once a xenobiotic ligand is recognized by AhR, the AhRligand is translocated into the nucleus and forms a complex with AhR nuclear translocator (ARNT) and several other nuclear receptor coactivators including nuclear receptor coactivator 7 (NCOA7), retinoblastoma protein, neural precursor cell expressed, developmentally down-regulated-8, and small ubiquitin related modifier-114,15. The AhR-coactivators complex binds to its recognition DNA sequence, the xenobiotic responsive element, which leads to transcriptional induction of TCDD-responsive genes such as cytochrome P4501A1(CYP1A1)16,17. NCOA7, also known as estrogen receptor associated protein 140, is involved in potentiating transcription activation of various nuclear receptors. NCOA7 interacts with estrogen receptor (ER) α, ER-β, peroxisome proliferating-activated receptors-γ, and retinoic acid receptor-α and mediates receptor signaling in specific target tissues18. For example, NCOA7 interacts with ER-β, which regulates estrogen-mediated brain functions during aging in mouse19. However, there are no reports about the effect of NCO7A on expression of AhR target gene and imflammatory cytokines in human lung epithelial cells. Our previous study revealed that NCOA7 was differentially expressed between normal and COPD lung tissues. Based on this finding, here we investigated whether NCO7A regulates the transcriptional activities of AhR target gene and inflammatory cytokines in human bronchial epithelial cells and human lung carcinoma cells stimulated by TCDD treatment in vitro.
Materials and Methods
1. RNA expression of NCOA7 from public database
To estimate the expression levels and to identify alternatively spliced transcripts, the RNA-Seq reads were mapped to the human genome using TopHat (version 1.3.3, http://tophat.cbcb.umd.edu/index.html). RNA-seq data were extracted from our previous experiment (GEO accession No. GSE57148). The transcript levels were calculated and the relative transcript abundances were measured in fragments per kilobase of exon per million fragments mapped (FPKM) using Cufflinks (http://cufflinks.cbcb.umd.edu/index.html).
2. Cell culture and chemical treatments
Human bronchial epithelial cell line (BEAS-2B) and human lung carcinoma cell line (A549) were maintained in Dulbecco's modified Eagle's medium supplemented with 10% heatinactivated fetal bovine serum and 1% streptomycin. Cells were grown at 37℃ in a 5% CO2 humidified incubator. Cells were treated in serum-free medium with various concentrations of TCDD (0, 0.15 nM, and 6.25 nM) in the presence of 1 nM of pCMV-NCOA7 and mock vector. TCDD was dissolved in dimethyl sulfoxide and stored at room temperature until use.
3. RNA extraction and cDNA synthesis
Total RNA was isolated using Qiagen RNA isolation kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions and quantified by measuring the absorbance at 260 nm. Thereafter, first-strand cDNA synthesis was performed by using the high-capacity cDNA reverse transcription (RT) kit (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's instructions. Briefly, 1.5 µg of total RNA from each sample was added to a mix of 2.0 µL 10× RT buffer, 0.8 µL 25× dNTP mix (100 mM), 2.0 µL 10× RT random primers, 1.0 µL MultiScribe reverse transcriptase, and added nuclease-free H2O. The final reaction mix was kept at 25℃ for 10 minutes, heated to 37℃ for 120 minutes, heated for 85℃ for 5 seconds, and finally cooled to 4℃.
4. Quantification by real-time polymerase chain reaction
Quantitative analysis of specific mRNA expression was performed by quantitative real-time polymerase chain reaction (PCR), by subjecting the resulting cDNA to PCR amplification using 96-well optical reaction plates in the ABI 7300 Real-Time PCR System (Applied Biosystems). Twenty-five-microliter reaction mix contained 0.1 µL of 10 µM forward primer and 0.1 µL of 10 µM reverse primer (40 nM final concentration of each primer), 12.5 µL of SYBR Green Universal Mastermix, 11.05 µL of nuclease-free H2O, and 1.25 µL of cDNA sample. Assay controls were incorporated onto the same plate, namely, notemplate controls to test for the contamination of any assay reagents. After sealing the plate with an optical adhesive cover, the thermo cycling conditions were initiated at 95℃ for 10 minutes, followed by 40 PCR cycles of denaturation at 95℃ for 15 seconds, and anneal/extension at 60℃ for 1 minute. Melting curve was performed by the end of each cycle to ascertain the specificity of the primers and the purity of the final PCR product. The sequences of the primers are listed in Table 1.
5. Plasmid construction
DNA fragment was generated by RT-PCR using a polymerase with proof reading activity (Pfu high fidelity polymerase; Invitrogen, Carlsbad, CA, USA) and the oligonucleotides introducing HindIII and XhoI sites in the fragment. The amplified PCR product was cloned into TOPO vector resulting in TOPO-NCOA7 and was introduced by transformation in Escherichia coli TOP10 cells. The insert was confirmed by sequencing. TOPO-NCOA7 was further digested with HindIII and XhoI restriction enzymes and purified by gel purification kit (Invitrogen). Purified TOPO-NCOA7 was ligated using T4 DNA ligase (Invitrogen) to obtain TOPO-NCOA7 construct. TOPO-NCOA7 was established by transformation in E. coli TOP10 cells.
6. Transient transfection
All transfections were performed in six-well plates after complexing the plasmid DNA with the Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. For each six-well plate, 2×105 A549 cells and 1×105 BEAS-2B cells were seeded in 2-mL media and incubated overnight. Before transfection, cells were washed with serum-free medium and resuspended in 0.2 mL Opti-MEM (Invitrogen). For transfection with Lipofectamine, an optimal ratio of DNA:lipid was used (1 µg, 4 µL for A549 and BEAS-2B cells). After incubated at 37℃ for 24 hours in Opti-MEM, then the media was replaced with complete cell media. pCMV-GFP was used as a transfection control.
Results
1. NCOA7 isoform 4 is highly expressed in COPD lung tissue compared to normal lung tissues
From the RNA-sequencing database, the RNA-sequencing results showed higher transcript levels of NCOA7 isoform 4 in COPD lung tissues compared to the normal control tissues (Figure 1).
2. Effect of NCOA7 isoform 4 on CYP1A1 mRNA expression
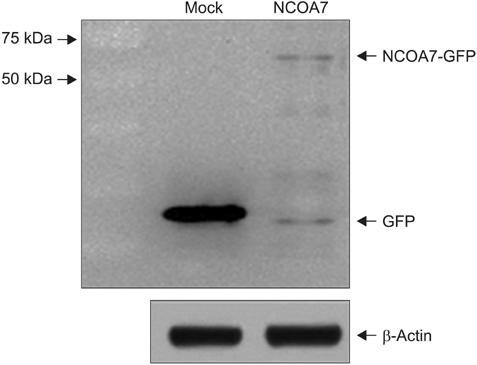
We examined the effect of NCOA7 isoform 4 on the transcriptional activity of CYP1A1 in BEAS-2B cells and A549 cells. BEAS-2B and A549 cells were transfected with NCOA7-GFP and GFP (as control) expression plasmids. NCOA7-GFP level was determined by Western blot analysis (Appendix 1). After transfection, the amount of CYP1A1 transcript was increased by treatment of either 0.15 or 0.65 nM TCDD in both cell lines. Expression level of CYP1A1 mRNA was significantly lower in NCOA7-transfected cells compared to the mock-transfected cells in BEAS-2B cell cultures treated with 0.65 nM TCDD (p<0.05) (Figure 2A). However, expression level of CYP1A1 mRNA was significantly higher in NCOA7-transfected cells compared to the mock-transfected cells in A549 cell cultures treated with 0.65 nM TCDD (p<0.05) (Figure 2B).
3. Effect of NCOA7 isoform 4 on pro-inflammatory cytokines
We next investigated the effect of NCOA7 isoform 4 on the transcriptional activity of pro-inflammatory cytokines in BEAS-2B cells and A549 cells. Based on the results from the multiple gene expression analysis (data not shown), the NCOA7 isoform 4-induced expression of IL-6 and IL-8 was investigated in further detail in addition to the expression of CYP1A1. In BEAS-2B cell cultures, the overexpression of NCOA7 isoform 4 did not change the level of IL-8 transcript but significantly decreased the transcriptional activity of IL-6 at 0.15 nM and 0.65 nM TCDD (Figure 3A, C). In contrast, the mRNA levels of IL-6 and IL-8 were significantly upregulated by NCOA7 isoform 4-transfected A549 cell cultures but not affected by TCDD treatment (p<0.05) (Figure 3B, D).
Discussion
Cigarette smoke is a powerful inducer of inflammatory responses20,21. Putative target genes of AhR are pro-inflammatory cytokines including IL-6 and IL-822,23. Previous report also showed that the AhR/ARNT pathway is important for the modulation of inflammatory mediators, including IL-6, IL-8, and cyclooxygenase-2, especially after stimulation by agents including dioxins24. Our previous work showed that NCOA7 isoform 4 was differentially expressed between normal and COPD lung tissues. Based on the result, in this study, we sought to examine the effect of NCOA7 isoform 4 on the expression of AhR target gene and pro-inflammatory cytokines in normal and abnormal lung cancer cell lines. Interestingly, TCDD treatment with the overexpression of NCOA7 isoform 4 in BEAS-2B and A549 cell cultures showed a different transcriptional activity of CYP1A1. Overexpression of NCOA7 isoform 4 exhibited an inhibitory effect on CYP1A1 expression in BEAS-2B cells, but promoted its expression in A549 cells. We also could observe similar expression patterns of pro-inflammatory cytokines (IL-6 and IL-8).
The induction of cell cycle activity and pro-inflammatory cytokines and increased activity of CYP1A1 enzyme contribute to the formation and growth of cancer cells25. The reversal contributes to maintain normal homeostasis. CYP1A1 plays a key role in the metabolism of both endogenous and exogenous substrates26,27,28. It has been reported that CYP1A1-mediated reactions result in the formation of reactive metabolites that is able to initiate carcinogenesis through DNA alterations and cell cycle modulation29. Rodriguez and Potter30 reported that CYP1A1 promotes the proliferation of breast cancer cells by increase of cyclin D1 and survivin. Since AhR directly mediates the increased expression of the CYP1A1 enzyme, the activation of AhR via environmental factors such as TCDD and formation of a complex with AhR coactivators including NCOA7 is associated with cancer initiation. Our result showed that overexpression of NCOA7 isoform 4 attenuated the transcriptional activity of CYP1A1 in normal cells (BEAS-2B) exposed to TCDD, which might be associated with mechanism of chemoprevention to maintain their normal homeostasis and avoid cancer initiation. On the contrary, overexpression of NCOA7 enhanced CYP1A1 expression in lung cancer cells (A549) exposed to TCDD, which might contribute to enhance growth of cancer via increased activity of CYP1A1 enzyme. These results suggest that NCOA7 isoform 4 may act as positive or negative regulator for AhR signaling pathway in normal and abnormal physiological conditions. In fact, AhR has dual roles in xenobiotic stimulations, in particular TCDD, and as a key player in normal physiological processes.
In conclusion, the present study indicates that NCOA7 isoform 4 has different regulatory role for AhR-TCDD signaling pathway in normal and cancer cells and might be therapeutic target for cancer treatment or management. Therefore, further studies will be needed to reveal the exact molecular mechanism of NCOA7 isoform 4 regarding to the AhR signaling pathway in both normal and cancer cells.



 XML Download
XML Download