

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Introduction
Lymphomatoid granulomatosis (LYG) is a rare Epstein-Barr virus (EBV) driven B-cell lymphoproliferative disorder. Atypical lymphoid cells directly accumulate within the affected tissues and clinically present in the form of infiltrative nodular lesions and with T-cell invasion and blood vessel destruction1. LYG most frequently involve the lung, but other common sites of involvement are the brain, kidney, liver, and skin2. Most present as multiple lung nodules, predominantly involving the lung bases, but occasionally atypical radiologic features can appear.
Here, we present a case of low grade pulmonary LYG with multiple lung lesions and endobronchial mass which is a rare presentation.
Case Report
A 54-year-old female was admitted with a 1-month history of dyspnea, cough and pulmonary infiltrates. She was a never smoker and had no preexisting lung disease. She was diagnosed with monoclonal gammopathy of unknown significance (MGUS) and autoimmune hemolytic anemia 4 years ago, and treated with cyclophosphamide previously.
On admission, her initial vital signs were stable and physical examination was unremarkable. Her white blood cell count was 6,860/L, hemoglobin was 8.7 mg/dL, and platelet count was 148,000/mm3. In blood chemistry, high sensitivity C-reactive protein (hs-CRP) was 0.32 mg/dL, lactate dehydrogenase levels increased to 1,238 U/L, total bilirubin was 1.90 mg/dL, and direct bilirubin was 0.53 mg/dL. During admission, the patient developed fever up to 39.7℃ with respiratory symptoms and hs-CRP increased to 2.06 mg/dL.
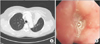
Initial chest radiograph showed diffuse patchy and nodular densities in the left lower lung (Figure 1A) and a low dose computed tomography of chest revealed variable sized round nodules and segmental collapse with proximal endobronchial filling defect in the left upper lingula segment (Figure 1B, C).
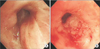
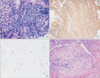
Bronchoscopic examination revealed diffuse bronchial nodular lesions with hypervascular mucosal changes and luminal narrowing at both bronchi (Figure 2A). Especially, left upper lobe (LUL) lingula division was almost nearly obstructed due to nodular lesion (Figure 2B). Endobronchial biopsy was performed at lingula division. Histopathologic examination showed infiltration with polymorphic atypical lymphoid cells and histiocytes (Figure 3A). Immunohistochemistry for CD3, CD20 as well as in situ hybridization for EBV were performed. Special stains demonstrated that the atypical population was characterized by many CD3-positive small reactive T cells (Figure 3B) with some scattered large CD20-positive B cells (Figure 3C). In situ hybridization for EBV-encoded RNA revealed a consistent number of EBV-positive large B cells (Figure 3D). This was consistent with a diagnosis of LYG grade 1.
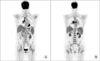
After corticosteroid pulse therapy, the patient's symptoms recovered and there was gradual but slow resolution of radiograph findings. After discharge, she was on maintenance corticosteroids. Nevertheless, after 8 months her respiratory symptoms recurred and computed tomography (CT) findings revealed LUL total collapse and follow-up bronchoscopy revealed total obstruction of LUL lingula segment (Figure 4). Cyclophosphamide therapy was started and 1 year later, there was complete regression of endobronchial tumor and atelectasis in LUL as well as multiple lung nodules (Figure 5). In spite of improvement in the lungs, 1 month later on, she developed enlargement of multiple lymph nodes in the neck, mediastinum and abdomen. Neck lymph node excisional biopsy was done and the result confirmed diffuse large B-cell lymphoma. Positron emission tomography CT results showed fludeoxyglucose uptakes in bilateral lower neck, mediastinum, peribronchial, mesentery and retroperitoneum nodes (Figure 6). The patient is receiving chemotherapy (R-CHOP) and is showing partial regression.
Discussion
LYG is a rare, EBV related lymphoproliferative disorder that is classified by the number of EBV-positive atypical B cells3. Age of onset is ~30-50 years and male seems to be more susceptible than females2. LYG is seen in various immunodeficiency states, such as acquired immune deficiency syndrome, various autoimmune diseases, post-transplantation immunodeficiency, and use of immunosuppressant medications1,4.
Nearly 90% of patients are symptomatic at diagnosis and present with a 4- to 8-month history of general and respiratory symptoms. Respiratory symptoms, mainly cough and dyspnea are found in more than half of cases. Other symptoms such as weight loss, sweating, acute respiratory distress may be present1,2,5. Extrapulmonary site of involvement mainly affect the skin, nervous system, and kidneys1.
The most common radiographic feature of LYG is multiple bilateral lung nodules of variable size involving mainly the lung base1. The lesions can progress rapidly, coalesce and commonly cavitate, therefore mimicking Wegner's granulomatosis or metastases6. In rare cases, it can present as a solitary large pleural based mass, idiopathic interstitial pneumonia or a lung abscess6,7. In our case, along with multiple lung nodules, there was also an endobronchial mass causing atelectasis, which is a rare presentation. There have only been two reports that describe of endobronchial involvement in previous literature7. However, our case is the only case to show complete regression of endobronchial mass and atelectasis after therapy. The endobronchial involvement was confirmed with bronchoscopic biopsy.
Histopathologic finding of LYG is an angiocentric polymorphous mononuclear infiltrate composed of numerous small lymphocytes, plasma cells, histiocytes and large EBV-postive B cells, which resemble immunoblasts. The small lymphocytes are mainly T cells, which are negative for EBV8. Based on the number of EBV-positive large B cells, LYG can be divided into 3 grades. Grade 1 contains only a few EBV-positive B cells, and grade 3 contains almost entirely of EBV-positive large B cells, and is regarded as an aggressive lymphoma9.
There has been no standard treatment established and the outcome of this disease is poor. In addition, patients often respond initially, but relapse is very common8. Patients with lower-grade lesions may be observed for regression, but patients with high-grade LYG require immediate treatment2. Therapies include corticosteroids, anti-CD20 monoclonal antibodies such as rituximab, interferon-alpha-2b and combination chemotherapy including cyclophosphamide. Corticosteroids are the most common treatment of LYG, but relapse is the rule and they are not useful for long-term disease control1.
The prognosis of LYG is variable but generally poor. Twenty percent of stage 1 patients can achieve spontaneous remission, but the disorder is often progressive10. Studies have shown a median survival of 14 months and a mortality of 65%-90%7. Many patients die as a result of pulmonary hemorrhage, central nervous system involvement, or transformation to an aggressive lymphoma. Our case was interesting as the lung lesions of stage 1 LYG that showed complete remission after therapy, still transformed to an aggressive lymphoma later on.
There have only been three cases of pulmonary lymphomatoid granulomatosis reported in Korea11,12,13. This is the first report of low grade LYG developed from a patient with MGUS receiving cyclophosphamide treatment. In our case there was an endobronchial mass as well as lung nodules, which is an unusual manifestation. Our case suggests that bronchoscopic examinations are needed to confirm endobronchial involvements in LYG. We also emphasize that in low grade LYG, where disease regression is noted after therapy, there is always the possibility to transform into malignant lymphoma. This should be considered when treating low grade LYG.


 XML Download
XML Download