

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Immunoglobulin G4-related disease (IgG4-RD) is a recently established systemic disease characterized by elevated serum IgG4 levels and marked lymphocyte and IgG4-positive plasma cell infiltration with fibrosis in involved organs1. It can affect virtually all organs, including the lung and pleura. Intrathoracic involvement of IgG4-RD has been reported by several investigators and shows various manifestations, including pulmonary nodules, round ground glass opacities (GGOs), mediastinal lymph node enlargement, and nodular pleural thickening2,3,4. Recently, interstitial lung disease such as nonspecific interstitial pneumonia (NSIP) was also reported to be another manifestation of lung involvement of IgG4-RD5,6. Here, we describe a case of IgG4-RD presenting as a form of interstitial lung disease.
Case Report
A 35-year-old man was seen in an outpatient clinic because of productive cough and dyspnea upon exertion persisting for 1 month. His sputum was yellowish in color and up to 150 mL daily in amount, and he had grade 3 dyspnea according to the Medical Research Council dyspnea scale. In addition to his chief complaint, he had had blood-tinged sputum intermittently during the previous 1.5 years. He did not report fever, night sweat, significant weight loss, or chest pain. He had had allergic rhinitis and bronchial asthma 1 year before presentation and intermittently used inhalers (salmeterol, fluticasone) and oral medications (montelukast, levocetirizine). He had never been a smoker. He was a software programmer for mobile phones and had a 10-month history of exposure to dust from a pottery factory that was located near to his workplace until 6 months prior to presentation. At presentation, he was alert and had an acutely ill appearance. His blood pressure was 125/75 mm Hg, heart rate was 79 beats/min, respiratory rate was 18 breaths/min, and body temperature was 36.3℃. Physical examination showed pinkish conjunctiva, anicteric sclera, and no palpable lymph nodes around the neck. The thorax showed a symmetric expansion without retraction and vesicular breath sounds were audible without crackle or wheezing on auscultation of the chest. The heart beat was regular and a cardiac murmur was not noted. Abdominal examination was unremarkable and there was no clubbing of the fingers and no skin rash.
A complete blood count showed a white blood cell count of 9,500/mm3 (61% neutrophils, 22% lymphocytes, and 8.6% eosinophils), hemoglobin of 15.7 g/dL, and a platelet count of 206,000/mm3. In serum biochemistry tests, the following values were found: calcium, 8.7 mg/dL; glucose, 95 mg/dL; creatinine, 0.88 mg/dL; total cholesterol, 185 mg/dL; protein, 8.8 g/dL; albumin, 4.7 g/dL; aspartate aminotransferase, 20 IU/L; alanine aminotransferase, 7 IU/L; alkaline phosphatase, 69 IU/L; and total bilirubin, 0.7 mg/dL. A chest radiograph showed bilateral subpleural irregularities and ill-defined GGOs and reticular opacities in both upper lung fields (Figure 1). Computed tomography (CT) of the chest revealed bilateral pleural thickening and subpleural fibrosis in both upper lobes, diffuse GGOs and bronchiectasis in the left upper lobe, and focal GGOs in the left lower and right middle lobes (Figure 2). Pulmonary function tests showed mild restrictive patterns with the following results: forced vital capacity (FVC), 72% of the predicted value (3.96 L); forced expiratory volume in 1 second (FEV1), 71% of the predicted value (3.30 L); FEV1/FVC ratio, 0.83; total lung capacity, 74% of the predicted value (5.57 L); and diffusing capacity for carbon monoxide, 88% of the predicted value (29.4 mL/mm Hg/min).
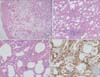
Considering the results of the chest CT and the pulmonary function test, interstitial lung disease, such as pleuroparenchymal fibroelastosis (PPFE) or chronic hypersensitivity pneumonitis (HP), was suspected. To rule out connective tissue disease as a cause of interstitial lung disease, serum autoantibodies were measured. Anti-SS-A/Ro antibodies were detected, but the patient did not complain of any rheumatologic symptoms. Bronchoalveolar lavage (BAL) was done in the left upper lobe and BAL fluid analysis showed an increase in eosinophils (12% of total white blood cells, 840/mm3). Bacteria, acid-fast bacilli, fungi, and viruses were not grown in the BAL fluid culture. Video-assisted thoracoscopic surgery was performed in the left upper lobe. Histological examination showed pleural and subpleural fibrosis with diffuse cellular infiltration throughout the alveolar wall. A specific distribution such as centrilobular or subpleural dominancy and granuloma were not seen. The cells consisted of lymphoplasma cells and mild lymphoid follicles were noted (Figure 3A-C). The elastic fiber in the pleura was not increased on elastic Van-Gieson staining. These microscopic findings did not fit into any categories of interstitial lung disease. Based on the diffuse lymphoplasmacytic infiltration with lymphoid follicles, IgG4-RD was suspected and immunohistochemical staining with IgG4 antibody (1:1,600, Binding Site, Birmingham, UK) was performed. IgG4-immunopositive cells were observed and the average number of these cells was 11 to 12 per high power field (HPF) (Figure 3D). However, storiform fibrosis and obliterative phlebitis and arteritis were not seen. Serum levels of IgG (1,930 mg/dL; normal range, 700-1,600 mg/dL) and IgG4 (270 mg/dL; normal range, 6.1-121.4 mg/dL) were also increased. We considered IgG4-related lung disease to be the most likely diagnosis. Involvement of organs other than lungs was not suspected clinically. We treated the patient with anti-inflammatory agents (prednisolone 0.5 mg/kg/day, azathioprine 2 mg/kg/day). His respiratory symptoms and chest radiograph have improved and he is under regular follow-up (Figure 4).
Discussion
IgG4-RD is a systemic fibroinflammatory condition that is characterized by specific histological findings of involved organs (lymphoplasma cell infiltration stained with IgG and IgG4, storiform fibrosis, and obliterative phlebitis), increased serum IgG4 levels, and a good response to steroids1. Discussion about IgG4-RD stemmed from elevated serum IgG4 concentrations in autoimmune pancreatitis (AIP) and extrapancreatic manifestations of AIP. IgG4-RD has been described in nearly all organs. According to one retrospective report on extrapancreatic manifestations in 90 patients with AIP, more than 90% of the patients showed extrapancreatic lesions4. The distribution and the frequency of involved organs were as follows: hilar lymph nodes of lungs (78%), bile duct (78%), peripancreatic or para-aortic lymph nodes (57%), lungs (54%), lacrimal or salivary glands (48%), retroperitoneum (20%), kidneys (14%), and prostate (10%). Although clinical manifestations of IgG4-RD vary depending on the organs involved, the histological features show marked similarities across the organs. Pathogenetic mechanisms in IgG4-RD are not yet clear. Potential triggering factors are certain bacterial infection and subsequent formation of antibodies acting on self antigens through a molecular mimicry mechanism or autoimmunity1. In genetically predisposed persons, these triggers provoke an immune reaction which is mediated predominantly by type 2 helper T (Th2) cells and activated regulatory T (Treg) cells in affected organs. High levels of Th2 cytokines, including interleukin (IL)-4, IL-5, and IL-13, as well as IL-10 and transforming growth factor β which are produced by activated Treg cells give rise to eosinophilia, elevated serum IgG4 and IgE levels, and characteristic infiltration of IgG4-positive plasma cells and fibrosis in the tissue. During this pathogenesis, the role of abundant IgG4 antibodies is not defined. Treatment of IgG4-RD is derived from treatment of AIP. Steroid therapy (prednisolone at a dose of 0.6 mg/kg/day) initially induces remission of the disease in the majority of patients1. However, without maintenance steroid therapy, relapses of the disease are common. Relapses occur within 3 years from the beginning of steroid therapy in most cases. Re-introduction of steroid therapy is as effective as initial therapy. Patients with recurrent or refractory disease in spite of steroid therapy can be treated with rituximab. Although the prognosis of IgG4-RD appears to be favorable with steroid therapy, massive infiltration of inflammatory cells and progressive fibrosis in IgG4-RD can cause serious dysfunction and failure of affected organs.
Since the first case of IgG4-related lung disease was reported in 20045, several cases have been reported in small sized case series. Zen et al.3 classified IgG4-related lung disease into four categories based on radiologic findings: the solid nodular type, the bronchovascular type, the alveolar interstitial type, and the round GGO type. Of these, the alveolar interstitial type, which is characterized by diffuse GGOs and reticular opacities, traction bronchiectasis, and honeycombing, resembles NSIP both radiologically and histologically. Few cases of this type were reported as a form of NSIP or usual interstitial pneumonia (UIP)6,7,8,9.
To our knowledge, our present case is the first Korean IgG4-RD patient presenting with interstitial lung disease. Although a total of 6 Korean cases of IgG4-related lung and pleural disease have been reported in the literature, these cases showed multiple lung nodules or masses10,11, consolidative masses12, or pleural nodules13,14 mimicking a malignancy. Our case showed relatively rapidly progressive (within 1 year) pleural and subpleural fibrosis with diffuse GGOs in both upper lobes, which is an uncommon presentation. Differential diagnoses included idiopathic interstitial pneumonia such as PPFE, inhalation lung injury, chronic HP, or inflammatory sequelae caused by pulmonary tuberculosis. Pulmonary tuberculosis was ruled out based on the negative culture and polymerase chain reaction results of BAL fluid for Mycobacterium tuberculosis, and the possibility of inhalation injury or HP seemed to be low because some typical findings such as peribronchial or centrilobular features, air trapping, or granuloma were not noted. Radiologic findings showing a predominant upper lobe distribution of the lesion were quite different from the usual NSIP findings, and an increase in elastic fiber characteristically seen in PPFE was not observed on histological examination. A strong suspicion of the possibility of IgG4-RD with diffuse lymphoplasmacytic infiltration on histology made diagnosis possible in this case.
Recently, expert consensus was reached for the diagnosis of IgG4-RD15. According to the guidelines, for probable diagnosis of IgG4-related lung disease with a surgical specimen, the following features are required: one (or at least two for highly suggestive diagnosis of IgG4-related lung disease) of the characteristic histological features-dense lymphoplasmacytic infiltrates, storiform fibrosis, or obliterative phlebitis-and a number of IgG4-positive plasma cells, which should be more than 50 cells/HPF and should account for more than 40% of IgG-positive plasma cells. However, IgG4-RD involving some organs, such as the lung or lymph nodes, may lack these histological features15. Storiform fibrosis or obliterative phlebitis might not be observed, particularly when IgG4-related lung disease presents as non-solid lesions, including interstitial pneumonia3. In addition, obliterative arteritis is identified mainly in solid lesions of IgG4-related lung disease.
Three cases of isolated IgG4-related interstitial lung disease have been reported. The first case, which was reported by Takato et al.6 in 2008, showed GGOs, reticular opacities with honeycombing, and traction bronchiectasis in both lower lobes on chest CT. Histological examination revealed diffuse lymphoplasmacytic infiltration and interstitial fibrosis with a homogenous distribution similar to that of NSIP. Numerous IgG4-positive plasma cells were noted. In the second case6, the patient's chest CT showed bilateral diffuse GGOs with traction bronchiectasis particularly in both lower lobes. Microscopically, infiltration of lymphocyte and plasma cells (mostly IgG4-positive plasma cells) was observed in the bronchovascular bundles and alveolar septa. The IgG4-to-IgG ratio of plasma cells reached 80%. Lastly, the case whose histological pattern was similar to that of UIP was recently reported9. Chest CT revealed bilateral pleural consolidations in the lower and upper lobes, diffuse GGOs, traction bronchiectasis, and small amounts of bilateral pleural effusions. On histological examination, features resembling UIP, such as diffuse interstitial fibrosis or honeycomb changes, were shown with interstitial lymphoplasmacytic infiltration. The average number of IgG4-positive plasma cells in the interstitium was more than 10/HPF. In all three cases, evidence of obliterative phlebitis was absent on histological examination, serum IgG4 levels were increased, and patients' symptoms and radiologic abnormalities improved significantly after treatment with steroids. In our case, storiform fibrosis, obliterative phlebitis, and obliterative arteritis were not seen and the number of IgG4-positive plasma cells did not reach the cut-off levels of the guidelines. However, given abnormally increased serum IgG4 levels and clinical improvement with steroids, this case is thought of to be IgG4-related lung disease.
Here, we report a case of IgG4-related lung disease showing a similar manifestation to interstitial lung disease. Because lung involvement of IgG4-RD can present with diverse clinical phenotypes and the treatment response to steroids is good, proper diagnosis of the disease and differential diagnosis from other interstitial lung disease are important. In conclusion, in a case of interstitial lung disease with diffuse lymphoplasmacytic infiltration, we should consider IgG4-related lung disease as a differential diagnosis.



 XML Download
XML Download