

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Knowledge of molecular pathogenesis of non-small cell lung cancer (NSCLC) has increased remarkably and changed principles of treatment especially for the past ten years. These changes are, however, limited mainly to adenocarcinoma of the lung. At this time, driver mutations can be identified in most cases of lung adenocarcinoma and targeted drugs like gefitinib, erlotinib, and crizotinib can be applied to about one third of the patients1.
Recently, genetic alterations in squamous cell lung cancer (SQCLC) have been studied extensively2 and are attracting attention because mutations, which have a critical role in the development of SQCLC and are frequently found in patients, can become targets of anti-cancer chemotherapy.
Go to : 
Clinical Characteristics of SQCLC
SQCLC now comprises about 30% of NSCLC but was the most common subtype of NSCLC in Korea. Patients with SQCLC usually complain cough, dyspnea, or hemoptysis. Tumors tend to be located in the central airways and are easy to form cavities. SQCLC is very strongly associated with smoking. Generally, SQCLC advances locally and shows fewer distant metastases than adenocarcinoma of the lung. In pathology, well-differentiated SQCLC characteristically shows keratinisation, intercellular bridges, and pearl formation. p63 is postitive and thyroid transcription factor 1 is negative on immunohistochemisty examination of SQCLC3.
Go to :
Pathogenesis of SQCLC
Pathogenesis of SQCLC seems to be different from that of adenocarcinoma, considering that there is a big difference in response to targeted therapies between the two types of NSCLC. Sequential pathogenesis of SQCLC is well known. Bronchial epithelial cells exposed to smoking for a long time develop to basal cell hyperplasia, squamous metaplasia, squamous dysplasia, carcinoma in situ, and finally squamous cell carcinoma. Together with these morphologic changes, chromosomal abnormalities, especially loss of heterozygosity, are accumulated during the process of carcinogenesis3.
Go to :
Genetic Alterations Found in SQCLC
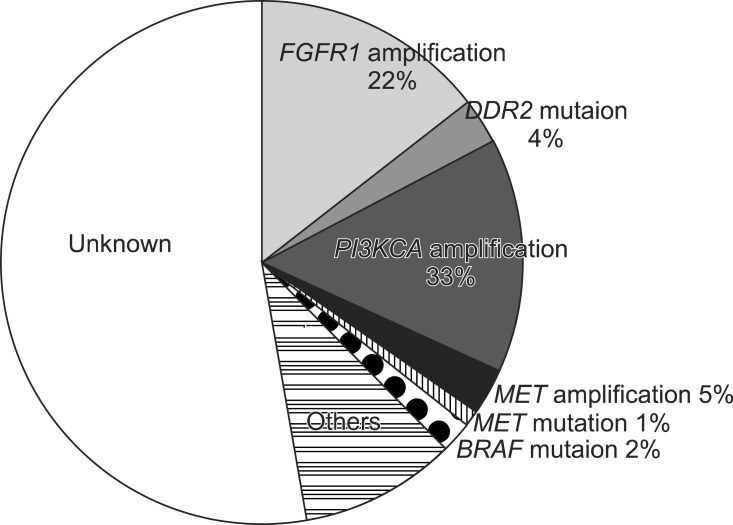
1. Membrane receptor alterations
1) Fibroblast growth factor receptor 1 (FGFR1) amplification
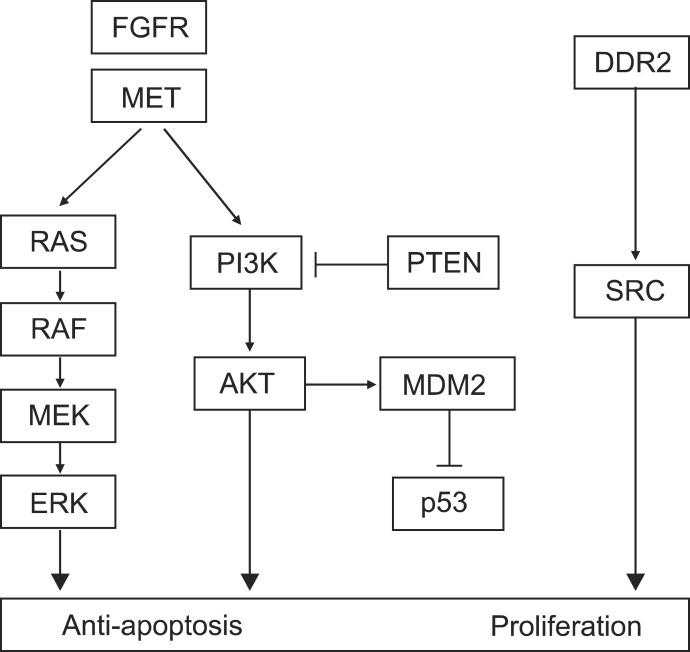
FGFR is a transmembrane receptor tyrosine kinase, involved in embryonal development, cell proliferation, cell differentiation, and regulation of angiogenesis. There are four kinds of members in FGFR family, and 22 fibroblast growth factor (FGF) ligands are known. It is known that activation of this pathway leads to enhanced growth of NSCLC cells (Figure 2) and that increased blood level and tissue expression of FGF is associated with a poor prognosis5.
Amplification of FGFR1, located in 8p12, was first reported as an important genetic change in SQCLC in 2010. Weiss et al.6 reported that 22% of 153 SQCLC tissues showed FGFR1 amplification by fluorescence in situ hybridization analysis. Dutt et al.7 identified FGFR1 amplification in 21% of 57 SQCLC samples and in 3% of adenocarcinomas of the lung by single nucleotide polymorphism array analysis. Both studies showed that growth of cell with FGFR1 amplification depends on FGFR1. Further, Weiss et al.6 showed treatment of PD173074, an inhibitor of FGFR, decreased tumor mass in a mouse model.
2) Discoidin domain receptor (DDR) 2 mutation
DDR, a receptor tyrosine kinase, plays a role in cell adhesion, proliferation, and extracellular remodeling after binding of collagen, an endogenous ligand (Figure 2). It was reported that upregulation of DDR1 was related to prolonged disease-free and overall survival of patients with NSCLC, especially SQCLC8.
Mutaions of DDR1 and DDR2, located in 1q23, have been found in SQCLC. Hammerman and coworkers reported that the frequency of DDR2 mutation was 3.8%, when they examined 290 SQCLC tissues and cell lines by DNA sequencing analysis. The mutations of DDR2 are activating mutations, and the activation was inhibited by treatment with dasatinib, a multi-kinase inhibitor. Further, they represented a SQCLC patient with a wild type EGFR who showed radiological response after treatment with both dasatinib and erlotinib in their clinical trial. The patient was confirmed to have a new mutation (S768R) in the DDR2 kinase domain9.
3) MET amplification
MET gene is located in 7q31 and expresses a receptor tyrosine kinase for hapatocyte growth factor. Overexpression of MET causes abnormal cell proliferation and invasion to neighboring tissues (Figure 2)10. Lung cancer cells with MET amplification are known to be very sensitive to crizotinib, an anaplastic lymphoma kinase/MET inhibitor. According to a previous report, MET amplification is more common in SQCLC than in other types of NSCLC and is associated with a poor prognosis10. Spiegel et al.11 reported that MetMab, a monoclonal antibody against MET, in combination with erlotinib showed a benefit in progression-free survival (2.9 months vs. 1.5 months, p=0.04) and overall survival (12.6 months vs. 3.8 months, p=0.002) compared with erlotinib alone in a MET-overexpressed subgroup of advanced NSCLC patients in a randomized phase II study.
2. Signaling pathway alterations
1) Phosphoinositide 3-kinase catalytic subunit α (PIK3CA) amplification and mutation
PIK3CA, located in 3q26, encodes a class I phosphoinositide 3-kinase (PI3K) α catalytic subunit (p110α). It is well known that PI3K-AKT pathway plays a critical role in survival and growth of diverse cancer cells (Figure 2)10. Both copy-number gain and mutations of PI3K are found in lung cancer. The copy-number gain of PI3K is found in 33.1% of SQCLC, in 6.2% of adenocarcinoma, and in 4.7% of small cell lung cancer. The mutations of PI3K are found in 6.5% of SQCLC and in 1.5% of adenocarcinoma4. Inhibitors of this pathway are in development and clinical trials using many kinds of PI3K inhibitors are ongoing for patients with SQCLC.
2) AKT1 mutation
E17K somatic mutation of AKT1, located in 14q32, activates the protein kinase continuously. This mutation is known to be present in about 1% of SQCLC but not in adenocarcinoma10. Akt inhibitors are currently being studied in several phase II trials.
3) Phosphatase and tensin homolog (PTEN) loss
PTEN, a tumor suppressor gene located in 10q23, encodes a lipid phosphatase inhibiting PI3K-AKT pathway and loss of PTEN activates PI3K-AKT signaling (Figure 2)10. Inactivation of PTEN by somatic PTEN deletions, mutations, and epigenetic mechanisms is found in many cancers. Reduction or loss of PTEN expression has been reported in up to 70% of NSCLC, both adenocarcinoma and SQCLC. PTEN mutations, occuring in approximately 5% of lung cancers, are significantly associated with squamous cell rather than adenocarcinoma histology (10.2% vs. 1.7%)3. Lung cancers with PTEN loss may be more sensitive to inhibitors of the PI3K pathway and clinical trials of PI3K inhibitors for cancers with PTEN loss are ongoing.
4) BRAF mutation
BRAF is a cytoplasmic serine/threonine kinase which plays an important role in the RAS-mitogen-activated protein kinase (MAPK) pathway (Figure 2). BRAF gene is located in 7q34. Mutation of BRAF causes increased kinase activity and consequently activates MAPK. The mutations of BRAF are found in ~2% of SQCLC and adenocarcinoma of the lung4. Selective inhibitors of BRAF are undergoing clinical trials now.
3. Transcriptional factor alterations
1) p53 mutation
p53, a tumor suppressor gene, is located in chromosome 17p13 and encodes a protein functioning mainly as a transcriptional factor which regulates the transcription of genes related to cell cycle arrest, apoptosis, and DNA repair. p53 mutations are found in more than half of NSCLC and in about 65% of SQCLC. Mutational hotspots are concentrated in the sequence-specific DNA-binding domain. Approximately 75% of mutations are missense mutations and lead to loss of function as a transcription factor. The mutations are affected by smoking. In a substantial number of tumors, wild-type p53 is inactivated by overexpression or amplification of MDM2 which ubiquinates p53 and marks it for degradation. Treatment with adenoviral vector was tried in the past but clinical trials using small molecules are now ongoing.
2) SOX2 amplification
Amplification of chromosome 3q26 is a kind of the most common genetic alterations found in SQCLC. SOX2 is a candidate oncogene present in this locus and amplification of SOX2 has been reported in about 20% of SQCLC. SOX2 is a transcriptional factor which plays an important role in regulation of stem cell function and development of lung epithelium10. Bass et al.12 showed that inhibition of SOX2 suppressed cell growth. But following studies confirmed that SOX2 amplification is not enough for carcinogenesis in itself and additional mutations of downstream effectors are needed to make a cancer. Clinical trials with SOX2 inhibitors are not ongoing.
4. Ongoing clinical trials using targeted drugs in SQCLC
Clinical trials of new therapies to some of the targets described are already underway for the treatment of SQCLC (Table 1). Those for FGFR1 are in progress most actively.
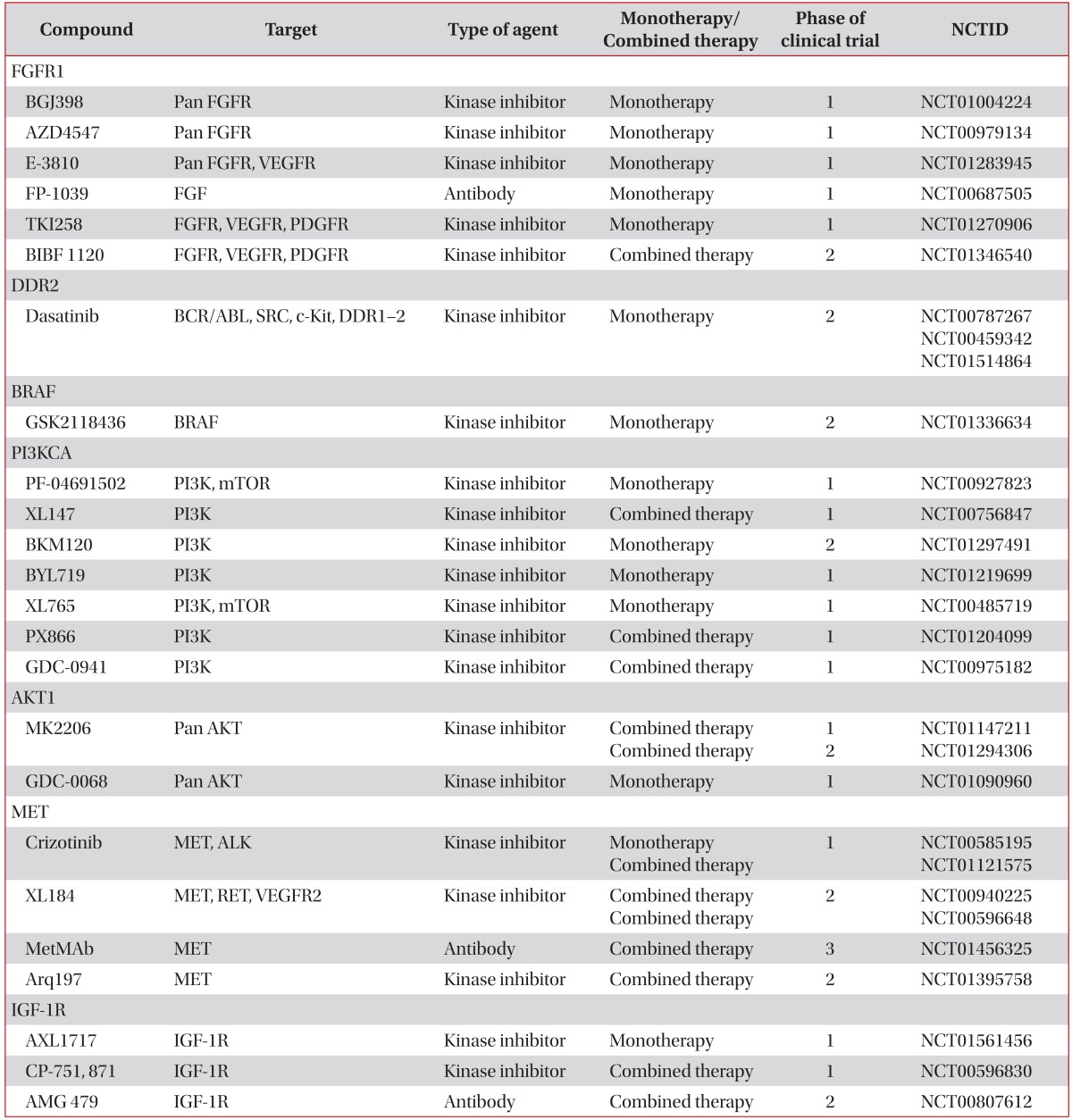
Table 1
Drugs targeting driver mutations in squamous cell lung cancer
NCTID: National Cancer Trial Identification; FGFR1: fibroblast growth factor receptor 1; VEGFR: vascular endothelial growth factor receptor; PDGFR: platelet-derived growth factors receptor; DDR2: discoidin domain receptor 2; PI3KCA: phosphoinositide 3-kinase catalytic subunit α; PI3K: phosphoinositide 3-kinase; mTOR: mammalian target of rapamycin; ALK: anaplastic lymphoma kinase; IGF: insulin-like growth factor.
![]()
Goss et al.13 are reported interim analysis of a phase II/III study of cediranib vs. placebo in combination with carboplatin and paclitaxel as initial therapy for 251 NSCLC patients. Cediranib (Recentin; AstraZeneca, Luton, UK) is an oral small molecule tyrosine inhibitor that inhibits vascular endothelial growth factor receptors (VEGFRs), platelet-derived growth factors receptors (PDGFRs), c-kit, and FGFR1. The result showed improved responses in the cediranib arm (38% vs. 16%, p=0.001), although this study was stopped due to excess toxicities, including hypertension, hypothyroidism, hand-foot syndrome, and gastrointestinal toxicity on the cediranib arm. A study with a lower dose of cediranib has been initiated.
Reck et al.14 reported a randomized phase II trial with BIBF 1120 (Boehringer Ingelheim, Ingelheim am Rhein, Germany), an oral small molecule inhibitor of FGFRs, PDGFRs, and VEGFRs. In their trial conducted in advanced NSCLC (n=73) after failure of platinum-based chemotherapy, 48% of patients treated with BIBF 1120 reached stable disease and the median progression-free survival was 6.9 weeks. Toxicities were manageable, with liver enzyme elevations, diarrhea, nausea, vomiting, and abdominal pain being the most common. Now, two phase III trials that compared BIBF1120 to placebo in addition to either docetaxel or pemetrexed as second-line treatments are underway.
Altorki et al.15 reported a multicenter, open-label, phase II "window of opportunity" trial with pazopanib (Votrient; GlaxoSmithKline, Brentford, UK), another oral small molecule inhibitor of the VEGFRs, PDGFRs, c-kit, and FGFRs. In the trial of 2 to 6 weeks of treatment with pazopanib in clinical stage I, II NSCLC (n=26), 86% of patients had some tumor reduction, with three partial responses. Pazopanib was well tolerated in this study, with the most common adverse events being hypertension, diarrhea, and fatigue. Phase II/III clinical trials of pazopanib in NSCLC are ongoing (NCT01208064, NCT00775307).
Go to :
Conclusion
Although adenocarcinoma is now the most common type of lung cancer, SQCLC still occupies a major part of lung cancer. However, targeted therapeutic drugs against SQCLC are not developed until now unfortunately. In case of adenocarcinoma, targeted therapy was developed through basic researches on carcinogenesis, translational researches, and clinical trials and molecular classification, as well as pathologic classification of cancers, became critical during this process. Recently, more and more genetic alterations of SQCLC have been identified and positive results of clinical trials using agents targeting these changes attract our attention.
Go to :
 XML Download
XML Download